हनटिंग्टन रोग
| Huntington's disease वर्गीकरण एवं बाह्य साधन | |
 | |
|---|---|
| A microscope image of Medium spiny neurons (yellow) with nuclear inclusions (orange), which occur as part of the disease process, image width 360 µm | |
| आईसीडी-१० | G10., F02.2 |
| आईसीडी-९ | 333.4, 294.1 |
| ओएमआईएम | 143100 |
| डिज़ीज़-डीबी | 6060 |
| मेडलाइन प्लस | 000770 |
| ईमेडिसिन | article/1150165 article/792600 article/289706 |
| एम.ईएसएच | D006816 |
हनटिंग्टन रोग, कोरिया, या विकार (HD), एक ऐसा तंत्रिका-अपजननात्मक आनुवंशिक विकार है जो मांसपेशियों के समन्वय को प्रभावित करता है और संज्ञानात्मक रोगह्रास और मनोभ्रंश की ओर ले जाता है। यह आम तौर पर अधेड़ अवस्था में दिखाई देने लगता है। HD कोरिया नामक असाधारण अनायास होने वाली छटपटाहट का अत्यधिक सामान्य आनुवंशिक कारण है और यह एशिया या अफ़्रीका की तुलना में पश्चिमी यूरोपीय मूल के लोगों में बहुत ज़्यादा आम है। यह रोग व्यक्ति के हनटिंग्टन नामक जीन की दो प्रतियों में से किसी एक पर अलिंगसूत्र संबंधी प्रबल उत्परिवर्तन द्वारा होता है, यानी इस रोग से पीड़ित माता-पिता के किसी भी बच्चे को वंशानुगत रूप से इस रोग को पाने का 50% ख़तरा रहता है। दुर्लभ स्थितियों में, जहां दोनों माता-पिता में एक प्रभावित जीन है, या माता-पिता में किसी एक की दो प्रतियां प्रभावित हैं, तो यह जोखिम काफी बढ़ जाता है। हनटिंग्टन रोग के शारीरिक लक्षण नवजात अवस्था से लेकर बुढ़ापे तक किसी भी उम्र में प्रकट हो सकते हैं, लेकिन आम तौर पर यह 35 और 44 साल की उम्र के बीच प्रकट होता है। लगभग 6% मामले अगतिक-अनम्य संलक्षण सहित 21 साल की उम्र से पहले शुरू हो जाते हैं; वे तेजी से बढ़ते हैं और इनमें थोड़ी भिन्नता होती है। इसके रूपांतरण को किशोर (जूवनाइल), अगतिक-अनम्य (अकाइनेटिक-रिजिड) या HD के वेस्टफ़ाल रूपांतरण के रूप में वर्गीकृत किया गया है।
हनटिंग्टन जीन सामान्य रूप से एक प्रोटीन के लिए आनुवांशिक जानकारी प्रदान करता है, जो "हनटिंग्टन" कहलाता है। हनटिंग्टन जीन कोड का उत्परिवर्तन एक अलग प्रकार के प्रोटीन रूप का संकेत देता है, जिसकी उपस्थिति से मस्तिष्क के विशिष्ट हिस्से क्रमिक रूप से क्षतिग्रस्त होने लगते है। यह किस तरीके से होता है इसे अब तक पूरी तरह नहीं समझा जा सका है। आनुवंशिक परीक्षण विकास के किसी भी चरण में कराई जा सकती है, लक्षण की शुरुआत से पहले भी. किस उम्र में व्यक्ति को इस परीक्षण के लिए परिपक्व माना जाय, अपने बच्चों की जांच कराने के प्रति माता-पिता के अधिकार और परीक्षण परिणामों की गोपनीयता और प्रकटीकरण के संबंध में यह अनेक नैतिक बहसों को जन्म देती है। आनुवंशिक परीक्षण करवाने पर विचार करने वाले व्यक्तियों की सूचना और सहायता के लिए आनुवांशिक परामर्श का विकास हुआ है और यह अन्य आनुवंशिक प्रबल रोगों के लिए एक आदर्श बन गया है।
रोग के लक्षण व्यक्तियों और एक ही परिवार के सदस्यों के बीच भी अलग-अलग हो सकते हैं, लेकिन अधिकांश लोगों में लक्षणों की प्रगति एक जैसी होती है। प्रारंभिक लक्षण हैं समन्वय की सामान्य रूप से कमी और एक अस्थिर चाल. रोग जैसे-जैसे बढ़ता है, वैसे-वैसे मानसिक क्षमताओं में ह्रास और व्यवहार तथा मनोविकार संबंधी समस्याओं के साथ-साथ बिना ताल-मेल वाली, झटकेदार शारीरिक गति प्रकट होने लगती है। शारीरिक क्षमताएं धीरे-धीरे तब तक अवरुद्ध हो जाती हैं जब तक कि समन्वित गति मुश्किल हो जाती है और मानसिक क्षमताओं का ह्रास होते हुए आम तौर पर वह मनोभ्रंश में बदल जाती है। न्यूमोनिया, हृदय रोग और गिरने से पहुंचने वाली शारीरिक चोट जैसी जटिलताएं जीवन की संभावना को लक्षणों के प्रारंभ होने के बाद लगभग बीस वर्षों तक समेट देती है। HD के लिए कोई इलाज नहीं है और बीमारी के बाद के चरणों में पूरे समय देखभाल की आवश्यकता होती है, लेकिन इसके लक्षणों में से कुछ के प्रति राहत देने के लिए उपचार सामने आ रहे हैं।
स्वयं-सहायक सहायता संगठन, जो पहले 1960 में स्थापित हुए और जिनकी संख्या में वृद्धि हो रही है, जनता में जागरूकता बढ़ाने, व्यक्तियों और उनके परिवार वालों को समर्थन देने तथा अनुसंधान को बढ़ावा देने के लिए काम कर रहे हैं। हेरिडिटरी डीज़िस फाउंडेशन, पहले समर्थक संगठन से जन्मे एक अनुसंधान समूह ने, 1993 में जीन ढूंढने में महत्वपूर्ण भूमिका निभाई थी। उसके बाद से कुछ वर्षों के अंतराल में महत्वपूर्ण खोज होती रही हैं और रोग की समझ में सुधार हो रहा है। वर्तमान शोध की दिशा में शामिल हैं रोग की वास्तविक क्रियाविधि का निर्धारण, अनुसंधान को तेजी से आगे बढ़ाने के लिए पशु नमूनों में सुधार, रोग के लक्षणों या धीमी प्रगति के उपचार के लिए दवाइयों के नैदानिक परीक्षण और रोग की वजह से होने वाली क्षति को सुधारने के लक्ष्य सहित वंश कोशिका रोगोपचार जैसी अध्ययन प्रक्रियाएं.
संकेत व लक्षण
हनटिंग्टन रोग के लक्षण सामान्यतः 35 और 44 वर्ष के बीच की आयु में प्रकट होने लगते हैं, लेकिन शैशव से लेकर किसी भी उम्र में हो सकते हैं।[1][2] प्रारंभिक चरणों में, व्यक्तित्व, संज्ञानता और शारीरिक कौशल में सूक्ष्म परिवर्तन हो सकते है।[1] आम तौर पर शारीरिक लक्षण पहले नज़र आते हैं, जबकि प्रारंभिक चरणों में संज्ञानात्मक और मनोविकार संबंधी लक्षण इतने प्रबल नहीं होते कि अलग से पहचाने जा सके.[1] हनटिंग्टन रोग से ग्रस्त लगभग हर रोगी अंततः एकसमान शारीरिक लक्षणों को दर्शाता है, लेकिन इसकी शुरूआत, प्रगति और संज्ञानात्मक तथा मनोविकार संबंधी लक्षणों की मात्रा में व्यक्तियों के बीच काफ़ी भिन्नता रहती है।[3][4]
सर्वाधिक विशेष प्रारंभिक शारीरिक लक्षण हैं कोरिया (लास्य) कहलाने वाली झटकेदार, अनियमित और अनियंत्रित चाल.[1] कोरिया शुरूआत में सामान्य बेचैनी, अनजाने में प्रवर्तित या असंपूर्ण छोटी गति, समन्वय की कमी, या धीमे झटकेदार नेत्र संचलन के रूप में प्रदर्शित होती है।[1] ये छोटी गतिजनक असामान्यताएं आम तौर पर कम से कम तीन वर्षों में गतिजनक शिथिलता के स्पष्ट संकेत के रूप में पहले दिखाई देती हैं।[3] विकार की प्रगति के साथ-साथ अकड़न, छटपटाहट सहित गति या असामान्य भंगिमा जैसे लक्षण स्पष्ट रूप से प्रकट होते हैं।[5] यह इसका संकेत हैं कि गति के लिए जिम्मेदार मस्तिष्क की प्रणाली प्रभावित हो गई है।[6] मनोप्रेरक कार्य इस प्रकार बहुत ज़्यादा बिगड़ती जाती है कि ऐसा हर कोई कार्य प्रभावित होता है, जिसमें मांसपेशी का नियंत्रण अपेक्षित हो. आम परिणाम हैं शारीरिक अस्थिरता, चेहरे के असामान्य हाव-भाव और चबाने, निगलने और बोलने में कठिनाई.[5] खाने में कठिनाइयों की वजह से आम तौर वज़न घट जाता है और इससे कुपोषण हो सकता है।[7][8] नींद की गड़बड़ी भी इससे जुड़े लक्षणों में शामिल है।[9] जुवेनाइल (किशोर) HD इन लक्षणों से थोड़ा अलग है जो आम तौर पर तेजी से पनपता है और यदि मौजूद हो, तो प्रमुख लक्षण के रूप में कुछ समय के लिए ऐंठन लिए हुए कोरिया प्रदर्शित होता है। जब्ती भी इस प्रकार के HD का आम लक्षण है।[5]
| चिड़चिड़ापन | 38-73% |
| उदासीनता | 34-76% |
| व्यग्रता | 34-61% |
| अवसादग्रस्त भावदशा | 33-69% |
| जुनूनी और बाध्यकारी | 10-52% |
| मनोविक्षिप्त | 3-11% |
संज्ञानात्मक क्षमताएं उत्तरोत्तर क्षतिग्रस्त होती हैं।[6] विशेष रूप से कार्यकारी प्रयोजन प्रभावित होते हैं जिनमें शामिल हैं आयोजना, संज्ञानात्मक नम्यता, अमूर्त चिंतन, आदेश अधिग्रहण, समुचित कार्य प्रवर्तन और अनुचित कार्यों की मनाही.[6] यह रोग जैसे-जैसे बढ़ता है, स्मृति का ह्रास की ओर झुकाव नज़र आने लगता है। प्राप्त रिपोर्टों के अनुसार ये अल्पकालीन स्मृति क्षय से लेकर दीर्घकालिक स्मृति की समस्याओं तक व्यापक रूप से फैली हुई हैं, जिनमें शामिल है प्रासंगिक (जीवन संबंधी स्मृति), कार्यविधिक (कार्य किस प्रकार निष्पादित किया जाए से संबंधित शरीर की स्मृति) और कार्यसाधक स्मृति का अभाव.[6] संज्ञानात्मक समस्याएं समय के साथ बिगड़ती जाती हैं, जो अंततः मनोभ्रंश में परिणत होती है।[6] क्षीणता के इस नमूने को मस्तिष्कप्रांतस्था संबंधी मनोभ्रंश कहा गया है, ताकि इसे अवप्रांतस्थीय मनोभ्रंश संलक्षण से अलग पहचाना जा सके, उदा. अल्ज़ाइमर रोग.[6]
रिपोर्ट किए गए तंत्रिका-मनोविकार के प्रकट रूप हैं व्यग्रता, अवसाद, भावनाओं की अभिव्यक्ति में कमी (भावशून्यता), अहंकेंद्रिता, आक्रामकता और बाध्यकारी व्यवहार, जो बाद में लत पैदा कर सकता है या और बिगाड़ सकता है जिसमें शामिल हैं मदिरापान, जुआ और अतिकामुकता.[10] दूसरे व्यक्ति के नकारात्मक भाव को पहचानने में परेशानियां भी देखी गई हैं।[6] अध्ययनों के बीच इन लक्षणों की व्यापकता में भी काफ़ी अस्थिरता पाई गई, जहां आजीवन मनोरोग विकारों के प्रसार के लिए अनुमानित दर 33% और 76% के बीच रहे हैं।[10] अनेक पीड़ितों और उनके परिजनों के लिए ये लक्षण इस रोग के सबसे दुखदायी पहलू हैं, जो अक्सर दैनंदिन क्रियाकलाप को प्रभावित करते हैं और संस्थाकरण के लिए कारण गठित करते हैं।[10] सामान्य लोगों की अपेक्षा इनमें आत्महत्या के विचार और आत्महत्या के प्रयास अधिक होते हैं।[1]
उत्परिवर्ती हनटिंग्टन सारे शरीर में व्यक्त होता है और प्रांतस्थ ऊतकों की विकृतियों से जुड़ा है जो सीधे मस्तिष्क के बाहर ऐसी अभिव्यक्ति के कारण होते हैं। इन विकृतियों में शामिल हैं मांसपेशी अपक्षय, हृदय विफलता, क्षतिग्रस्त ग्लूकोस सह्यता, वज़न घटना, अस्थि-सुषिरता और वृषण क्षीणता.[11]
आनुवांशिकी
प्रत्येक मनुष्य में हनटिंग्टिन जीन (HTT) होता है, जो हनटिंग्टिन प्रोटीन (Htt) के लिए कूटबद्ध होता है। इस जीन का एक अंश ट्राइन्यूक्लियोटाइड आवृत्ति नामक आवृत्ति अनुभाग है, जिसकी लंबाई प्रत्येक व्यक्ति में अलग होती है और पीढ़ियों के बीच लंबाई में परिवर्तन हो सकता है। जब इस पुनरावृत्त अनुभाग की लंबाई किसी निश्चित सीमा तक पहुंचती है, तो यह उत्परिवर्ती हनटिंग्टन प्रोटीन (mHtt) नामक प्रोटीन के एक परिवर्तित रूप का उत्पादन करता है। इन प्रोटीनों के भिन्न कार्य ही रोगात्मक परिवर्तनों के कारण होते हैं, जो बदले में रोग के लक्षण पैदा करते हैं। हनटिंग्टन रोग उत्परिवर्तन आनुवंशिक रूप से प्रबल और लगभग पूर्णतः अंतर्वेधित है: व्यक्ति के किसी एक HTT जीन का उत्परिवर्तन रोग का कारण बनता है। यह लिंग के अनुसार नहीं, बल्कि जीन के पुनरावृत्त भाग की लंबाई के अनुसार वंशागत होता है और इसकी गंभीरता का भी यही कारण है, प्रभावित माता-पिता के लिंग से प्रेरित किया जा सकता है।[12]
आनुवंशिक उत्परिवर्तन
HD कई ट्राइन्यूक्लियोटाइड आवृत्ति विकारों में से एक है, जो सामान्य विस्तार से अधिक जीन के पुनरावृत्त भाग की लंबाई से उत्पन्न होता है।[13] HTT जीन 4p16.3 पर गुणसूत्र 4[13] की छोटी भुजा पर स्थित होता है। HTT में तीन DNA क्षारकों का अनुक्रम शामिल होता है – साइटोसिन-एडिनिन-ग्वॉनिन (CAG)-जिनकी कई बार पुनरावृत्ति होती है (अर्थात्. ... CAGCAGCAG...), जो ट्राइन्यूक्लियोटाइड आवृत्ति कहलाता है।[13] CAG एमिनो एसिड ग्लूटमाइन का आनुवंशिक कूट है, अत: इनकी एक श्रृंखला पॉलीग्लुटमाइन पथ (या polyQ ट्रैक्ट) नामक ग्लूटमाइन की श्रृंखला और जीन के पुनरावृत्त अंश, PolyQ भाग का उत्पादन करता है।[14]
| [13] | ||
| आवृत्ति संख्या | वर्गीकरण | रोग की स्थिति |
|---|---|---|
| <28 | सामान्य | अप्रभावित |
| 28-35 | मध्यम | अप्रभावित |
| 36-40 | कम अंतर्वेधन | + / - प्रभावित |
| > 40 | पूर्ण अंतर्वेधन | प्रभावित |
आम तौर पर, लोगों के पॉलीक्यू क्षेत्र में 36 पुनरावृत्त ग्लूटमाइन से कम ग्लूटमाइन होते हैं जो साइटोप्लास्मिक प्रोटीन हनटिंग्टन के उत्पादन में परिणत होता है।[13] तथापि, 36 या अधिक ग्लूटमाइन की श्रृंखला के परिणामस्वरूप एक ऐसे प्रोटीन का निर्माण होता है जिसकी विशेषताएं अलग होती हैं।[13] mHtt (उत्परिवर्ती Htt) नामक यह परिवर्तित प्रकार, कुछ न्यूरॉन के क्षय दर को बढ़ाता है। मस्तिष्क के हिस्सों में इस प्रकार के न्यूरॉनों की विभिन्न मात्रा और निर्भरता रहती है और तदनुसार ये प्रभावित होते हैं।[5] सामान्यतया, CAG पुनरावृत्ति की संख्या इस बात पर निर्भर करती है कि यह प्रक्रिया कितनी प्रभावित हुई है और लगभग 60% तक लक्षणों के आरंभ काल की विविधता का कारण बनती है। शेष विविधता के लिए परिवेश और HD क्रियाविधि को बदलने वाले अन्य जीन को उत्तरदायी ठहराया जाता है।[13] 36-40 पुनरावृत्ति रोग के कम-अंतर्वेधी रूप में परिणत होती है, जिसमें लक्षणों की देर से शुरूआत और धीमी प्रगति होती है। कुछ मामलों में प्रारंभ इतनी देरी से होता है कि लक्षणों को कभी देखा ही नहीं जाता.[15] बहुत बड़ी पुनरावृत्ति गणनाओं सहित, HD में पूर्ण अंतर्वेधन होता है और यह 20 से कम उम्र में होता है, जब उसे जुवेनाइल HD, अकाइनेटिक-रिजिड या HD का वेस्टफ़ाल रूपांतरण कहा जाता है। यह HD वाहकों के लगभग 7% के लिए जिम्मेदार है।[16]
आनुवंशिकता
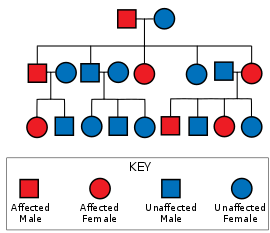
हनटिंग्टन रोग में अलिंगसूत्री प्रबल आनुवंशिकता है, यानी प्रभावित व्यक्ति विशेष रूप से अपने माता या पिता से विस्तृत ट्राइन्यूक्लियोटाइड आवृत्ति (उत्परिवर्ती युग्मजीविकल्पी) सहित जीन की एक प्रति प्राप्त करता है।[1] चूंकि उत्परिवर्तन का अंतर्वेधन अत्यंत उच्च होता है जीन की उत्परिवर्ती प्रति वाला व्यक्ति रोग से ग्रस्त होता है। इस प्रकार के वंशानुक्रम नमूने में, किसी प्रभावित व्यक्ति की प्रत्येक संतान को उत्परिवर्ती युग्मजीविकल्पी के वंशागत होने और इसलिए विकार से प्रभावित होने का 50% जोखिम रहता है (चित्र देखें). यह संभाव्यता लिंग-मुक्त है।[17]
28 से अधिक की ट्राइन्यूक्लियोटाइड CAG आवृत्तियां प्रतिकृति के दौरान असंतुलित होती हैं और यह असंतुलन मौजूद पुनरावृत्ति की संख्या के साथ बढ़ती रहती है।[15] यह प्रायः ट्राइन्यूक्लियोटाइड आवृत्ति की सटीक प्रति उत्पन्न करने के बजाय, पीढ़ियों के गुज़रने के साथ-साथ (गतिशील उत्परिवर्तन) नए विस्तार की ओर अग्रसर होता है।[13] इसके कारण क्रमागत पीढ़ियों में पुनरावृत्ति की संख्या में इस तरह परिवर्तन होता है कि "मध्यवर्ती" संख्या (28-35), या "कम अंतर्वेधन" (36-40) सहित कोई अप्रभावित माता या पिता पूर्णतः अंतर्वेधी HD उत्पन्न करने वाली पुनरावृत्ति की संख्या में वृद्धि सहित जीन की प्रति को आगे बढ़ा सकते हैं।[13] क्रमागत पीढ़ियों में पुनरावृत्ति की संख्या में इस प्रकार की वृद्धि (और फलस्वरूप रोग का शीघ्र प्रारंभ काल तथा गंभीरता आनुवांशिक प्रत्याशा कहलाती है।[13] शुक्र-जनन में असंतुलनता अंडाणु-जनन की अपेक्षा अधिक होती है;[13] मातृ पक्ष से वंशागत युग्मजीविकल्पी आम तौर पर एकसमान पुनरावृत्ति लंबाई के होते हैं, जबकि पितृ पक्ष से वंशागत युग्मजीविकल्पी की लंबाई बढ़ने की संभावना अधिक होती है।[13][18] ऐसे नए उत्परिवर्तन द्वारा हनटिंग्टन रोग का उत्पन्न होना दुलर्भ है, जहां माता या पिता किसी में 36 से अधिक CAG पुनरावृत्ति मौजूद नहीं हैं।[19]
बड़े समरक्त परिवारों के अतिरिक्त, दोनों जीन से प्रभावित व्यक्ति बहुत कम होते हैं।[20] कुछ समय के लिए HD को ही केवल एक ऐसा रोग माना गया जिसमें दूसरे उत्परिवर्ती जीन की मौजूदगी लक्षणों और विकास को प्रभावित नहीं करती,[21] परंतु उसके बाद पाया गया है कि यह समलक्षण और विकास की दर को प्रभावित कर सकती है।[13][20] किसी ऐसे व्यक्ति की संतान जिसमें दो प्रभावी जीन हों, उनमें से एक को वंशानुक्रम में पाता है और इसलिए निश्चित रूप से रोग को वंशागत प्राप्त करता है। ऐसे माता-पिता की संतान को दो प्रभावित जीन को वंशानुक्रम में पाने के 25% जोखिम सहित, वंशागत HD का 75% जोखिम रहता है।[17] समरूप जुड़वां में, जिन्होंने एकसमान प्रभावित जीन को वंशानुक्रम में प्राप्त किया है, आम तौर पर प्रारंभ काल और लक्षणों में भिन्नता रहती है।[20]
क्रिया-विधि
Htt प्रोटीन 100 से ज़्यादा अन्य प्रोटीनों के साथ परस्पर क्रिया करता है और इसमें एकाधिक जैविक कार्यों की मौजूदगी प्रतीत होती है।[22] उत्परिवर्ती mHtt प्रोटीन के व्यवहार को पूरी तरह नहीं समझा गया है, परंतु यह कुछेक प्रकार की कोशिकाओं के लिए विषाक्त है, विशेष रूप से मस्तिष्क के लिए. मुख्यतः क्षति स्ट्रिएटम को पहुंचती है, लेकिन जैसे-जैसे रोग बढ़ता है, मस्तिष्क के अन्य हिस्से भी पर्याप्त रूप से प्रभावित होते हैं। क्षति के जमा होने पर, मस्तिष्क के इन भागों के कार्यों से जुड़े लक्षण प्रकट होते हैं। गति की योजना और नियंत्रण स्ट्रिएटम के मुख्य कार्य हैं और इनमें परेशानियां प्रारंभिक लक्षण हैं।[12]
Htt कार्य
Htt सभी स्तनधारी कोशिकाओं में प्रकट होता है। यकृत, हृदय और फेफड़ों में सामान्य मात्रा सहित सर्वाधिक संकेंद्रन मस्तिष्क और अंडकोष में पाया जाता है।[12] मनुष्यों में Htt के कार्य अस्पष्ट हैं। यह उन प्रोटीनों के साथ परस्पर क्रिया करता है जो प्रतिलेखन, कोशिका संकेतन और अन्त:कोशिका अभिगमन में शामिल होता है।[12][23] पशुओं में HD प्रदर्शित करने के लिए आनुवांशिक रूप से परिवर्तित, Htt के अनेक कार्य पाए गए हैं।[24] इन पशुओं में, Htt भ्रूणीय विकास के लिए महत्वुपूर्ण होता है, क्योंकि इसका अभाव भ्रूणीय हत्या जुड़ी होती है। यह क्रमादेशित कोशिका मृत्यु की रोकथाम करते हुए एंटी-एपोप्टोटिक एजेंट के रूप में भी कार्य करता है और मस्तिष्क व्युत्पन्न न्यूरोट्रॉफ़िक कारक के उत्पादन को नियंत्रित करता है, जोकि एक ऐसा प्रोटीन है जो न्यूरॉन की रक्षा करता है और तंत्रिकाजनन के दौरान उसके निर्माण को नियमित करता है। Htt वायुकोशीय अभिगमन और अंतर्ग्रंथीय संचार को भी सुविधाजनक बनाता है और तंत्रिकाकोशिकीय जीन प्रतिलेखन को नियंत्रित करता है।[24] अगर Htt का प्रकटन बढ़ता है और अधिक Htt उत्पादित होता है, तो मस्तिष्क कोशिका अनुजीवन में सुधार होता है और mHtt के प्रभाव कम होते हैं, जबकि Htt का प्रकटन कम होने पर, परिणामी लक्षण mHtt की मौजूदगी की अभिलक्षक हैं।[24] मनुष्यों में सामान्य जीन का व्यवधान रोग पैदा नहीं करता.[12] संप्रति यह निष्कर्ष निकाला गया है कि रोग Htt के अपर्याप्त उत्पादन द्वारा पैदा नहीं होता, बल्कि mHtt के विषाक्त कार्य की प्राप्ति द्वारा होता है।[12]
mHtt के कारण कोशिकीय परिवर्तन
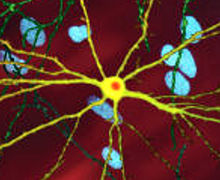
कई ऐसे कोशिकीय परिवर्तन हैं जिनके माध्यम से mHtt की विषाक्त क्रिया प्रकट होती है और HD विकृति उत्पन्न करती है।[25][26] mHtt के प्रतिलेखनोत्तर संशोधन की जैविक प्रक्रिया के दौरान, प्रोटीन की दरार पॉलीग्लूटमाइन विस्तार के कुछ भागों से बने छोटे अंशों को पीछे छोड़ सकती हैं।[25] जब ग्लूटमाइन में Htt प्रोटीन अत्यधिक मात्रा में होती है तो ग्लूटमाइन की ध्रुवीय प्रकृति, अन्य प्रोटीनों के साथ अन्तःक्रिया उत्पन्न करती है। इस प्रकार, Htt अणु तत्व एक दूसरे के साथ हाइड्रोजन बंध बनाते हैं, जिससे क्रियाशील प्रोटीन की वलित होने की जगह प्रोटीन समुच्चय बनता है।[27] समय के साथ, ये समूह इकट्ठा होते हैं, अंततः न्यूरॉन क्रिया में दखल देते हैं क्योंकि तब ये अंश प्रोटीन समुच्चयन नाम की प्रक्रिया में खुल कर और संगठित होकर, कोशिकाओं के भीतर समावेशी पिंड बनाते हैं।[25][27] न्यूरोनीय समावेश अप्रत्यक्ष हस्तक्षेप चलाते हैं। अतिरिक्त प्रोटीन समुच्चय न्यूरॉन में तंत्रिकाक्ष और पार्श्वतंतुओं पर एक साथ ढेर में जमा होते हैं, जो यांत्रिक रूप से न्यूरोट्रांस्मीटरों के संचार को बंद कर देता है, क्योंकि पुटिकाएं (न्यूरोट्रांस्मीटरों से भरी) कोशिका-कंकाल के माध्यम से नहीं गुज़र सकती. अंततः, समय के साथ, जैसे-जैसे न्यूरोनीय समावेश बढ़ते हैं, दूसरे न्यूरॉन्स को संकेत देने के लिए उपलब्ध न्यूरोट्रांस्मीटर कमतर होते जाते हैं।[27] समावेशी पिंड, कोशिकीय नाभिक और कोशिकाद्रव्य, दोनों में पाए गए हैं।[25] मस्तिष्कीय कोशिकाओं में समावेशी पिंड, सबसे पहले होने वाले विकृत परिवर्तनों में से एक हैं, तथा कुछ प्रयोगों ने यह दर्शाया है कि वे कोशिकाओं के लिए विषाक्त हो सकते हैं, लेकिन अन्य प्रयोग यह दर्शाते हैं कि ये शरीर के सुरक्षा तंत्र का हिस्सा बन सकते हैं और कोशिकाओं की रक्षा में सहायक हो सकते हैं।[25]
कई पथों की पहचान की गई है जिनके द्वारा mHtt कोशिका की मृत्यु का कारण बन सकता है। इनमे शामिल हैं: संरक्षिका प्रोटीन पर प्रभाव, जो प्रोटीन को बिखरने और बिखरे हुए प्रोटीन को हटाने में सहायक होते हैं; कास्पासेस के साथ अन्तःक्रिया, जो कोशिकाओं को हटाने की प्रक्रिया में भूमिका निभाते हैं; तंत्रिका कोशिकाओं पर ग्लूटमाइन का विषाक्त प्रभाव; कोशिकाओं के भीतर ऊर्जोत्पादन की क्षति; और जीन के प्रकटन पर प्रभाव. रेस नामक प्रोटीन के साथ अन्तःक्रिया के कारण mHtt का कोशिकाओं पर विषाक्त प्रभाव बड़ी मजबूती से बढ़ताहैं, जो मुख्यतः स्ट्रिएटम में प्रकट होता है।[28] पाया गया कि रेस mHtt के सूमोयलेशन को प्रेरित करती है, जो प्रोटीन पिंड को तोड़ देती है-कोशिका संवर्धन अध्ययनों ने जताया कि असमुच्चित रूप से पिंड कम विषाक्त हैं।[28]
एक अतिरिक्त सिद्धांत के अनुसार, जो HD द्वारा कोशिका के कार्यों को बाधित करने के तरीके को समझाता है, स्ट्रेटिएटल कोशिकाओं में माईटोकोंड्रिया की क्षति (माईटोकोंड्रियल कमी के कई वर्णन पाए गए) और न्यूरॉन में कई प्रोटीन के साथ हनटिंग्टिन प्रोटीन की अन्तःक्रिया, ग्लूटमाइन की अतिसंवेदनशीलता को बढ़ावा देती है, जो बहुत अधिक मात्रा में, एक्साइटोटॉक्सिन के रूप में पाई गई। एक्साइटोटॉक्सिन अनेक कोशिकीय संरचनाओं को क्षति पहुंचा सकता है। हालांकि ग्लूटमाइन अत्यधिक उच्च मात्रा में नहीं पाई गई, तथापि यह माना गया है कि वर्धित अतिसंवेदनशीलता के कारण, ग्लूटमाइन की सामान्य मात्रा भी एक्साइटोटॉक्सिन उत्पन्न कर सकती है। इसके अलावा, कैपासेस पर संवेदनशीलता की निर्भरता में वृद्धि, पॉलीग्लूटमाइन के आवृत्ति विस्तार और संवेदनशीलता में वृद्धि द्वारा सक्रिय होती हैं। हनटिंग्टिन प्रोटीन, कैपासेस द्वारा छोटे टुकड़ों में चीर दी जाती है; ये परमाणु समुच्चय, प्रतिलेखन को, न्यूरॉन के नाभिक में "दाखिल" होते हुए, प्रोटीन के निर्माण में दखल देकर बाधित करते हैं।[29][30] दुर्भाग्यवश, हस्तक्षेप की वजह से होने वाला कोशिकीय तनाव, एपॉप्टोसिस के होने तक और अधिक हनटिंग्टिन की टूटन को बढ़ावा देता हैं।[29]
mHtt के कारण सूक्ष्मदर्शी परिवर्तन

HD मस्तिष्क के विशिष्ट क्षेत्रों को प्रभावित करता है। सबसे प्रमुख प्रारंभिक प्रभाव नियोस्ट्रिएटम कहलाने वाले आधारिक गंडिकाओं के हिस्सों पर पड़ता है और जो पुच्छल नाभिक और कवच से निर्मित होता है।[12] अन्य प्रभावित क्षेत्रों में शामिल हैं श्याम द्रव्य, प्रमस्तिष्कीय वल्क की तीसरी, पांचवी और छठी परत, हिप्पोकैम्पस, अनुमस्तिष्क में पुरकिंजे कोशिकाएं, अधःश्चेतक की पार्श्विक नलाकार नाभिक और चेतक के हिस्से.[13] ये अपने भीतर शामिल न्यूरॉन और अपनी संरचना के हिसाब से प्रभावित होते हैं, जैसे-जैसे वे कोशिकाएं खोते जाते हैं उनका आकार छोटा होता जाता है। इन क्षेत्रों में होते हैं वे न्यूरॉन में से एक हैं प्रभावित अनुसार प्रकार और उनकी संरचना, आकार में कम करने के रूप में वे कक्षों खो देते हैं।[13] स्ट्रिएटल कांटेदार न्यूरॉन सबसे असुरक्षित हैं, विशेषकर बाह्य ग्लोबस पैलिडस की ओर झुकाव वाले, जिनके अंतःन्यूरॉन और भीतरी पैलीडम पर झुकी कांटेदार कोशिकाएं कम प्रभावित होती हैं।[13][31] HD एस्ट्रोसाइटों में भी असामान्य वृद्धि का कारण बनता है।[32]
आधारिक गंडिका-HD द्वारा सर्वाधिक प्रमुखता से प्रभावित मस्तिष्क का भाग-गति और व्यवहारपरक नियंत्रण में अहम भूमिका निभाता है। उनके कार्यों को पूरी तरह नहीं समझा गया है, लेकिन मौजूदा सिद्धांत प्रस्तावित करते हैं कि वे संज्ञानात्मक कार्यकारी प्रणाली[6] और मोटर सर्किट का हिस्सा हैं।[33] आधारिक गंडिका आम तौर पर उन परिपथों को बाधित करती है जो विशिष्ट गति उत्पन्न करते हैं। किसी विशिष्ट हरकत को आरंभ करने के लिए, प्रमस्तिष्कीय वाह्य संरचना द्वारा आधारिक गंडिका को संकेत भेजा जाता है जो अवरोध जारी करने का कारण बनता है। आधारिक गंडिका को पहुंची क्षति निषेध की रिहाई तथा पुनर्स्थापना को अनियमित और अनियंत्रित कर सकती है, जो चाल की अजीब शुरुआत या अनजाने में चाल की शुरुआत या अपने निर्दिष्ट समापन के पहले या बाद में गति को रोक सकता है। इस क्षेत्र में पहुंच रही कुल क्षति HD से जुड़ी लाक्षणिक अनियत चाल को अंजाम देती है।[33]
आधारिक गंडिका को दो प्रकार से क्षतिग्रस्त किया जा सकता है: प्रत्यक्ष और परोक्ष रूप से. सीधे मार्ग में, कम न्यूरोट्रांस्मीटरों को आंतरिक ग्लोबस पैलीडस (IGP) भेजे जाते हैं, जो इसे अवरोध में कमी के रूप में समाविष्ट कर लेते है, जिसके द्वारा सामान्य से अधिक न्यूरोट्रांस्मीटर मुक्त होते हैं। चेतक, जो असंख्य न्यूरोट्रांस्मीटर प्राप्त करते हैं, निरोधक हो जाते हैं और इस प्रकार गतिजनक प्रांतस्था को कम न्यूरोट्रांस्मीटर भेजता है। अंततः, गतिजनक प्रांतस्था कम उत्तेजित होती है और गति सामान्य से कम होती है। अप्रत्यक्ष मार्ग बाह्य ग्लोबस पैलीडस द्वारा कम न्यूरोट्रांसमीटर की प्राप्ति से शुरू होता है और बदले में, इसके प्रति प्रतिक्रया कम हो जाती है क्योंकि कम प्रतिरोध वाला संकेत अधिक न्यूरोट्रांस्मीटर जारी करता है। उपचेतक नाभिक (STN), जो बाह्य ग्लोबस पैलीडस से संकेत प्राप्त करता है, IGP को, प्राप्त अधिक न्यूरोट्रांस्मीटर के बदले कम न्यूरोट्रांस्मीटर जारी करता है। अब IGP काफी निरोधक होते हैं क्योंकि STN का कार्य IGP को उत्तेजित करना हैं और इसलिए IGP कम न्यूरोट्रांस्मीटर जारी करता है। इस सन्दर्भ में, चेतक द्वारा कम न्यूरोट्रांस्मीटर अभिग्रहण को कम अवरोध माना जाता है। अंत में, गतिजनक प्रांतस्था अधिक न्यूरोट्रांस्मीटर प्राप्त करती है और अतिउत्तेजित हो जाती है, जिससे झटकेदार हरकतें पैदा होती है, जो कोरिया में सामान्य है। क्योंकि स्ट्राएटम में दो भिन्न प्रकार के न्यूरॉन होते हैं, एक भिन्न प्रकार का न्यूरॉन, जो अलग तंत्रिकाक्ष और पार्श्वतंतु से लक्षित होता है, प्रत्येक मार्ग में उत्तेजित होता है (हालांकि दोनों में न्यूरोट्रांस्मीटर GABA प्रयुक्त होता है) और इस प्रकार दोनों एक साथ चल सकते हैं। आम तौर पर अप्रत्यक्ष मार्ग पहले प्रभावित होता है, जिसके कारण कोरिया पहले लक्षणों में से एक है, लेकिन अंततः, दोनों प्रकार के न्यूरॉन मर जाते हैं और गति काफी सीमित हो जाती हैं।[29]
प्रतिलेखनीय अनियंत्रण
CREB-बाध्यकारी प्रोटीन (CBP), एक प्रतिलेखन कारक, कोशिकाओं के कार्य करने के लिए महत्वपूर्ण है, क्योंकि सह-उत्प्रेरक के रूप में प्रवर्तकों की सार्थक संख्या में यह अस्तित्व मार्ग के लिए जीन का प्रतिलेखन शुरू करते हैं।[30] इसके अलावा, CBP का निर्माण करने वाले अमीनो एसिड में एक स्ट्रिप 18 ग्लूटामाइन शामिल है। इस प्रकार, CBP पर ग्लूटमाइन सीधे Htt श्रृंखला पर वर्धित संख्या के साथ ग्लूटमाइन से अन्तःक्रिया करता है और CBP अपनी नाभिक के पास की आम स्थिति से हटा दिया जाता है।[34] विशेष रूप से, CRB में एक असीटलट्रांस्फ़रेस कार्यक्षेत्र शामिल होता है, जिसने स्टेफ़न और साथियों द्वारा किए गए एक प्रयोग में यह दर्शाया कि CBP में 51 ग्लूटमाइनों के साथ Htt एक्सन 1 इस कार्यक्षेत्र से बंधे हैं।[30] उन व्यक्तियों के ऑटोप्सी किए गए मस्तिष्कों में भी, जिन्हें हनटिंग्टन रोग था, CBP की अविश्वसनीय मात्रा पाई गई।[34] इसके अतिरिक्त, जब CBP अधिक प्रकट होता है, पॉलीग्लूटमाइन से प्रेरित मौत कम होती हैं, जो आगे यह दर्शाता है कि CBP सामान्य रूप से हनटिंग्टन रोग और न्यूरॉन में एक महत्वपूर्ण भूमिका निभाता है।[30]
रोग-निदान
HD के प्रारंभ होने का चिकित्सीय निदान, रोग के विशेष शारीरिक लक्षणों के प्रकट होने के पश्चात किया जा सकता है।[1] परिवार में HD का कोई इतिहास उपलब्ध न हो तो शारीरिक निदान की पुष्टि करने के लिए आनुवंशिक परीक्षण का उपयोग किया जा सकता है। रोगलक्षणों के प्रारंभ होने से पहले भी, आनुवंशिक परीक्षण यह पुष्टि कर सकता है कि कोई व्यक्ति या भ्रूण, रोग उत्पन्न करने वाले HTT जीन में ट्राइन्यूक्लियोटाइड पुनरावृति की एक विस्तारित प्रति का वहन कर रहा है या नहीं. संपूर्ण जांच प्रक्रिया के दौरान सलाह एवं मार्गदर्शन प्रदान करने के लिए एवं सुनिश्चित निदान के निहितार्थों के संबंध में आनुवंशिक परामर्श उपलब्ध रहता है। इन निहितार्थों में शामिल हैं व्यक्ति की मनोवृत्ति, जीवन-वृत्ति, परिवार नियोजन संबंधी निर्णय, रिश्तेदारों एवं संबंधों पर पड़ने वाले प्रभाव. पूर्व-रोगलक्षण संबंधी परीक्षण की उपलब्धता के बावजूद, HD का वंशागत जोखिम वाले केवल 5% व्यक्ति परीक्षण करवाना पसंद करते हैं।[12]
नैदानिक
[[चित्र:Huntington.jpg|thumb|alt=Cross section of a brain showing undulating tissues with gaps between them, there are two large gaps evenly spaced about the centre|पुच्छल नाभिक के शीर्ष का अपक्षय, पार्श्विक निलयों के लालाटिक श्रृंग का परिवर्धन और सामान्यकृत मस्तिष्क-प्रांतस्था शोष दर्शाता HD ग्रस्त रोगी के MR ब्रेन स्कैन से किरीटी अनुभागसन्दर्भ त्रुटि: <ref> टैग के लिए समाप्ति </ref> टैग नहीं मिला[35] कंप्यूटरीकृत टोमोग्राफी (CT) एवं चुंबकीय अनुनाद इमेजिंग (MRI) जैसे चिकित्सापरक प्रतिबिंबन केवल रोग की विकसित अवस्थाओं में दृश्य प्रमस्तिष्कीय अपक्षय प्रदर्शित करते हैं। fMRI एवं PET जैसे कार्यात्मक तंत्रिका-प्रतिबिंबन तकनीक शारीरिक रोगलक्षणों के प्रारंभ होने से पहले मस्तिष्क की गतिविधि में परिवर्तन दर्शा सकते हैं।[13]
आनुवंशिकता
चूंकि HD प्रबल होता है, इसे ग्रहण करने की जोखिम वाले व्यक्तियों में निदान प्राप्त करने की एक तीव्र प्रेरणा होती है। HD के लिए आनुवंशिक परीक्षण में एक रक्त परीक्षण शामिल होता है जो प्रत्येक HTT युग्मजीविकल्पी के CAG पुनरावृत्तियों की संख्या की गणना करता है।[36] एक अनुकूल परिणाम को निदान नहीं माना जाता है, क्योंकि इसे रोगलक्षणों की शुरूआत से दशकों पहले ही प्राप्त किया जा सकता है। तथापि, एक नकारात्मक परीक्षण का अर्थ है कि व्यक्ति जीन की विस्तारित प्रति को वहन नहीं करता है एवं HD का विकास नहीं होगा.[13]
रोगलक्षण-पूर्व परीक्षण जीवन-परिवर्तक घटना और बहुत ही व्यक्तिगत निर्णय है।[13] HD के लिए परीक्षण का चयन करने के लिए प्रस्तुत मुख्य तर्क जीवन वृत्ति एवं पारिवारिक निर्णयों में सहायता प्रदान करना है।[13] HD वंशागत जोखिम वाले 95% से अधिक व्यक्ति परीक्षण नहीं करवाते हैं, अधिकांशतः इसलिए कोई इलाज मौजूद नहीं है।[13] एक महत्वपूर्ण मुद्दा व्यक्ति द्वारा अनुभव की जाने वाली चिंता है जो सकारात्मक परिणाम के प्रभाव की तुलना में अंततः उनमें HD विकसित होने की संभावना के बारे में जानकारी के अभाव से जुड़ा है।[12] परिणामों का लिहाज किए बिना, परीक्षण के दो वर्षों बाद तनाव स्तर पहले से कम पाए गए हैं, लेकिन अनुकूल जांच परिणाम के बाद आत्महत्या का जोखिम बढ़ जाता है।[12] इस विकार को वंशागत न ग्रहण करने वाले व्यक्ति प्रभावित होने वाले परिवार के सदस्यों के संबंध में उत्तरजीवी अपराध भाव का अनुभव कर सकते हैं।[12] परीक्षण पर विचार करते समय ध्यान में रखे गए अन्य कारकों में शामिल हैं भेदभाव की संभावना एवं अनुकूल परिणाम के निहितार्थ, जिसका मतलब आम तौर पर माता-पिता में एक प्रभावित जीन की मौजूदगी है और उस व्यक्ति के सहोदरों को वंशागत रूप में उसे ग्रहण करने का जोखिम हो सकता है।[12] HD में आनुवंशिक परामर्श प्रारंभिक निर्णय लेने के लिए और बाद में, चयन करने पर, संपूर्ण परीक्षण प्रक्रिया के सभी चरणों में, सूचना, सलाह एवं सहयोग प्रदान कर सकता है।[37] HD के लिए आनुवंशिक परीक्षण के उपयोग के संबंध में परामर्श एवं मार्गदर्शन अलिंगसूत्र प्रबल अनुमस्तिष्कीय गतिभंग जैसे अन्य आनुवंशिक विकारों के लिए आदर्श बन चुके हैं।[12][38][39] HD के लिए रोगलक्षण-पूर्व परीक्षण ने बहुपुटीय गुर्दा रोग, पारिवारिक अल्ज़ाइमर रोग एवं स्तन कैंसर जैसी आनुवंशिक रूपांतरण वाली अन्य बीमारियों के लिए परीक्षण को भी प्रभावित किया है।[38]
भ्रूणीय
कृत्रिम गर्भाधान के प्रयोग द्वारा उत्पन्न भ्रूणों का, आरोपण-पूर्व आनुवंशिक निदान के इस्तेमाल से HD के लिए आनुवंशिक परीक्षण किया जा सकता है। इस तकनीक का, जिसमें एकल कोशिका को 4 से 8 कोशिका वाले भ्रूण से निकाला जाता है और फिर आनुवंशिक असामान्यता के लिये उसकी जांच की जाती है, बाद में यह सुनिश्चित करने के लिए उपयोग किया जा सकता है कि प्रभावित HTT जीन वाले भ्रूणों का आरोपण न किया जाए और इससे किसी भी संतान में यह रोग प्रवेश नहीं कर पाएगा. गर्भाशय में विकसित होने वाले भ्रूण या विकसित हो चुके भ्रूण का प्रसवपूर्व रोग-निदान भी संभव है।[40]
सापेक्ष निदान
HD उत्पन्न करने वाले विस्तारित ट्राइन्यूक्लियोटाइड पुनरावृत्ति का पता करने के लिए विशिष्ट रोगलक्षणों पर आधारित लगभग 90% HD निदान एवं रोग संबंधी पारिवारिक इतिहास की पुष्टि आनुवंशिक परीक्षण द्वारा की जाती है। शेष अधिकांश रोगों HD सदृश विकार कहा जाता है।[5][41] अधिकांश इन अन्य विकारों को सामूहिक रूप से HD-सदृश (HDL) नाम दिया गया है।[41] अधिकांश HDL रोगों के कारण अज्ञात हैं, लेकिन ज्ञात कारणों वाले रोग प्रायन प्रॊटीन जीन (HDL 1), जंक्टोफिलिन 3 जीन (HDL2), एक अप्रभावी रूप से वंशागत HTT जीन (HDL3– जो केवल एक परिवार में पाया गया एवं बहुत कम जाना गया), तथा TATA बॉक्स-बाइंडिंग प्रोटीन (HDL4/SCA17) का संकेतन करने वाले जीन में उत्परिवर्तन के कारण होता है।[41] अन्य अलिंगसूत्री प्रबल रोग, जिनका HD के रूप में गलत निदान किया जा सकता है, डेंटाटोरूब्रल-पैलिडोलुइसियन अपक्षय एवं न्यूरोफ़ेरिटिनोपैथी हैं।[41] कुछ अलिंगसूत्री अप्रभावी विकार भी हैं जो HD के छिटपुट मामलों के समान दिखते हैं। मुख्य उदाहरण हैं कोरिया अकैंथोसाइटोसिस, पैंटोथिनेट काइनेस-संबंधी तंत्रिका अपजनन एवं X-संबद्ध मॅकलियॉड संलक्षण.[41]
प्रबंधन
HD का कोई इलाज नहीं है, लेकिन इसके लक्षणों में कुछ की गंभीरता को कम करने के उपचार उपलब्ध हैं।[42] इन उपचारों में से कई के लिए, विशेष रूप से HD के लक्षणों के उपचार में उनकी प्रभावशीलता की पुष्टि करने के लिए व्यापक नैदानिक परीक्षण अपूर्ण हैं।[43][44] जैसे-जैसे इस रोग की प्रगति होती है और व्यक्ति में अपनी जरूरतों को पूरा करने की क्षमता घटती है, सावधानीपूर्वक संचालित बहुविषयक देखरेख सेवा उत्तरोत्तर जरूरी होता जाता है।[43]
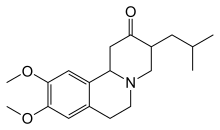
HD में कोरिया की तीव्रता को कम करने के लिए टेट्राबेन्ज़ीन को विशेष रूप से विकसित किया गया था,[43] 2008 में जिसके प्रयोग को अमेरीका में स्वीकृति मिली थी।[45] कोरिया को कम करने में सहायक अन्य दवाओं में न्यूरोलेप्टिक और बेन्जोडियाज़ेपाइन शामिल हैं।[2] अमन्टाडाइन या रेमासेमाइड जैसे यौगिक अभी भी परीक्षणाधीन हैं, लेकिन प्रारंभिक सकारात्मक परिणाम दिखा रहे हैं।[46] हाइपोकाइनेसिया और जड़ता का इलाज पार्किन्सन-रोधी दवाओं से किया जा सकता है और मायोक्लोनिक हाइपरकाइनेसिया का इलाज वैल्पोरिक एसिड से किया जा सकता है।[2]
मनोरोग लक्षणों का उपचार, सामान्य जनता के इलाज के लिए प्रयुक्त दवाओं से किया जा सकता है।[43][44] अवसाद के लिए चुनिंदा सेरोटोनिन पुनरुग्रहण प्रतिरोधकों और मिर्टाज़पाइन की सिफारिश की जाती है, जबकि मानसिक और व्यवहार संबंधी समस्याओं के लिए असामान्य मनोविकार-रोधी दवाओं की सिफारिश की जाती है।[44]
निगलने में कठिनाई के कारण वज़न घटने और खाने में होने वाली परेशानियां और अन्य मांसपेशीय सामंजस्य की समस्या आम हैं, जो बीमारी के बढ़ने के साथ ही पोषण प्रबंधन को उत्तरोत्तर महत्वपूर्ण बनाता जाता है।[43] प्रगाढक कारकों को तरल में मिलाया जा सकता है, क्योंकि गाढे तरल पदार्थ निगलने में आसान और सुरक्षित है।[43] रोगी को धीरे-धीरे खाने और मुंह में भोजन के छोटे कौर लेने के लिए याद दिलाते रहना चाहिए, यह खाने के रास्ते को अवरुद्ध होने से बचाने में सहायक होता है।[43] अगर भोजन करना अत्यधिक जोखिम भरा या असहज हो जाए तो त्वचाप्रवेशी गुहांतदर्शी जठरछिद्रीकरण के उपयोग का विकल्प उपलब्ध है। यह आहार नलिका है, जो स्थाई रूप से उदर से जुड़ते हुए पेट के भीतर जाती है, जो श्वास में अड़चन डालने वाले आहार के खतरे को कम करती है और बेहतर पोषण प्रबंधन प्रदान करती है।[47]
यद्यपि HD के संज्ञानात्मक लक्षणों के पुनर्वास में मदद करने में सहायक व्यायाम और चिकित्सा के लिए अपेक्षाकृत कुछ कम अध्ययन किए गए हैं, तथापि शारीरिक चिकित्सा, अभिग्रहण चिकित्सा और वाक चिकित्सा की उपयोगिता के कुछ सबूत मिलते हैं। फिर भी, स्वास्थ्य अधिकारियों द्वारा इन्हें और समर्थन करने के लिए अधिक परिशुद्ध अध्ययन आवश्यक हैं।[48] अशक्तता को सीमित करने के लिए एक बहुविषयक दृष्टिकोण महत्वपूर्ण हो सकता है।[49] उन व्यक्तियों के परिवारों को, जिन्हें HD वंशानुक्रम में मिली है या मिलने का जोखिम है, HD का कई पीढ़ियों का अनुभव होता है जो पुराना हो सकता है और इसमें हाल की सफलताओं और आनुवंशिक परीक्षण में सुधार, परिवार नियोजन विकल्प, देखभाल प्रबंधन और अन्य विचारों का अभाव हो सकता है। अपने ज्ञान को अद्यतन करने, अपने मिथकों को दूर करने और उनके भावी विकल्पों और योजनाओं पर विचार द्वारा उनकी सहायता करने पर केंद्रित, आनुवांशिक परामर्श इन व्यक्तियों को लाभ देता है।[12][50]
पूर्वानुमान
ट्राइन्यूक्लियोटाइड आवृत्ति की लंबाई, शुरूआती अवधि में बदलाव और लक्षणों की प्रगति की दर के 60% के लिए जिम्मेदार होती है। एक लंबे दोहराव के परिणामस्वरूप शीघ्र शुरूआती अवधि और लक्षणों की तेजी से प्रगति होती है।[13][51] उदाहरण के लिए, लोगों में ट्राइन्यूक्लियोटाइड आवृत्ति के 60 से अधिक दोहराव अक्सर बीस वर्ष की आयु से पहले ही बीमारी का विकास कर देता है और ट्राइन्यूक्लियोटाइड का 40 से कम दोहराव सुस्पष्ट लक्षण विकसित नहीं करता है।[52] शेष परिवर्तन पर्यावरणीय कारकों और अन्य जीन के कारण होते हैं जोकि बीमारी की प्रक्रिया को प्रभावित करते हैं।[13]
लक्षणों के दिखने की शुरूआत से HD में जीवन प्रत्याशा सामान्यतः लगभग बीस वर्ष है।[5] जीवन के लिए जोखिमपूर्ण अधिकांश जटिलताएं मांसपेशीय समन्वय मुद्दों से पैदा होती हैं और कुछ हद तक व्यवहार में परिवर्तनों के कारण, जो संज्ञानात्मक क्रियाओं से पैदा होती हैं। HD से पीड़ित लोगों में एक तिहाई लोगों की मृत्यु निमोनिया के कारण होती है, जो इसका सबसे बड़ा जोखिम है। जैसे-जैसे तालमेल वाली गतिविधियों में कमी आती जाती है, फेफड़ों को साफ़ करने में कठिनाई और खाने या पीने से श्वास लेने में कठिनाई के वर्धित जोखिम के कारण निमोनिया का खतरा बढ़ जाता है। दूसरा सबसे बड़ा जोखिम हृदय रोग है, जो लगभग एक चौथाई HD के रोगियों की मृत्यु का कारण बनता है।[5] मौत का अगला सबसे बड़ा कारण आत्महत्या है, जिसकी वजह से लगभग 7.3% HD पीड़ित लोग अपनी जान ले लेते हैं, जबकि लगभग 27% ऐसा करने का प्रयास करते हैं। यह स्पष्ट नहीं है कि आत्महत्या के विचार किस हद तक मनोवैज्ञानिक लक्षणों से प्रभावित होते हैं, क्योंकि इसे एक व्यक्ति द्वारा अपने जीवन पर नियंत्रण रखने की भावना को बनाए रखने या बीमारी की अंतिम अवस्था से बचने की प्रतिक्रिया माना जा सकता है।[53][54][55] घुटन, गिरने से लगने वाली शारीरिक चोट और कुपोषण अन्य संबद्ध जोखिम हैं।[5]
जानपदिक रोग-विज्ञान
हनटिंगटन रोग की देर से शुरूआत का मतलब है कि यह आम तौर पर प्रजनन को प्रभावित नहीं करता.[12] दुनिया भर में HD का प्रचलन 5-10 व्यक्ति प्रति लाख जनसंख्या है,[56][57] लेकिन जातीयता, स्थानीय प्रवास और विगत आप्रवास पैटर्न के परिणामस्वरूप भौगोलिक दृष्टि से ये भिन्न होते हैं।[12] इसका प्रसार पुरुषों और महिलाओं में एकसमान है। इसकी मौजूदगी का दर पश्चिमी यूरोपीय मूल के लोगों में सबसे अधिक है, जिसका औसत सत्तर व्यक्ति प्रति लाख लोग के आसपास है और दुनिया के बाकी हिस्सों में कम है, उदाहरण के लिए एशियाई और अफ्रीकी मूल के लोगों में यह प्रति मिलियन एक है।[12] इसके अतिरिक्त, कुछ स्थानीयकृत क्षेत्रों में इसका औसत अपने क्षेत्रीय औसत से बहुत अधिक है।[12] वेनेज़ुएला के माराकैबू झील क्षेत्र की पृथक आबादी में इसके प्रचलन की दर सर्वाधिक दरों में से है, जहां HD प्रति मिलियन सात हजार लोगों को प्रभावित करता है।[12][58] उच्च स्थानीयकरण के अन्य क्षेत्र तस्मानिया और स्कॉटलैंड, वेल्स तथा स्वीडन के विशिष्ट क्षेत्रों में पाए गए हैं।[55] कुछ मामलों में वर्धित प्रचलन स्थानीय संस्थापक प्रभाव, भौगोलिक अलगाव वाले क्षेत्र में वाहकों के ऐतिहासिक प्रवास के कारण होता है।[55][59] इनमें से कुछ वाहकों का पता वंशावली अध्ययनों के उपयोग द्वारा, सैकड़ों वर्ष पीछे जा कर लगाया है।[55] आनुवंशिक आवृत्ति लोप भी, इसकी उपस्थिति के लिए भौगोलिक विविधताओं के सुराग़ दे सकता है।[55][60]
आनुवंशिक परीक्षण की खोज तक, आंकड़े केवल शारीरिक लक्षणों, HD के पारिवारिक इतिहास पर आधारित नैदानिक निदान शामिल कर सकते थे, जिनमें उन लोगो को अलग कर दिया जाता था जिनकी मृत्यु लक्षण प्रकट होने से पहले किन्ही अन्य कारणों से हुई हो. इन मामलों को अब आंकड़ों में शामिल किया जा सकता है तथा जैसे-जैसे परीक्षण और अधिक व्यापक रूप से उपलब्ध हो जाता है, व्यापकता के अनुमान और विकार के होने में वृद्धि होने की संभावना रहती है।[55][61]
इतिहास
[[चित्र:On Chorea with photo.jpg|thumb|alt=On the right is a young man, dressed in suit and tie, sporting a moustache and tuft of hair on the chin; on the left is the top half of a medical journal titled 'Medical and Surgical Reporter' |1872 में जॉर्ज हनटिंग्टन ने 22 वर्ष की उम्र में अपने पहले "ऑन कोरिया" दस्तावेज़ में विकार का वर्णन किया।सन्दर्भ त्रुटि: <ref> टैग के लिए समाप्ति </ref> टैग नहीं मिला 1846 में चार्ल्स गोरमैन ने देखा कि किस प्रकार स्थानबद्ध प्रदेशों में उच्च व्याप्ति होती नज़र आती है।[62] जेफ़रसन मेडिकल कॉलेज के डंगलिसन के दोनों छात्र,[63] गोरमैन एवं वाटर्स से स्वतंत्र, जोहान क्रिश्चन लुंड ने 1860 में एक आरंभिक विवरण भी प्रस्तुत किया।[62] उसने विशेष रूप से नोट किया कि नॉर्वे के एक एकांत क्षेत्र सेटेस्डालेन में, परिवारों में प्रचलित प्रतिक्षेपक गति विकारों के एक स्वरूप से जुड़ी मनोभ्रंश की उच्च व्यापकता थी।[64]
रोग का प्रथम संपूर्ण विवरण 1872 में जॉर्ज हनटिंगटन द्वारा प्रस्तुत किया गया. समान रोगलक्षण प्रदर्शित करने वाले एक परिवार की कई पीढियों के सम्मिलित चिकित्सा इतिहास का परीक्षण करते हुए, उन्होंने समझा कि उनकी अवस्थाएं जुड़ी हुई होनी चाहिए; उन्होंने रोग की विस्तृत एवं सटीक परिभाषा को अपने प्रथम दस्तावेज़ के रूप में प्रस्तुत किया। अनजाने में, हनटिंग्टन ने मेंडेलीय वंशानुक्रम के पुनरन्वेषण के बरसों पहले, एक अलिंगसूत्री प्रबल रोग के वंशानुक्रम के सटीक स्वरूप का वर्णन किया। "अपने वंशानुगत स्वभाव के बारे में. कब माता-पिता में से किसी एक या दोनों ने रोग के प्रकटीकरण को प्रदर्शित किया है।.., एक या अधिक संतान प्राय: निरपवाद रूप से इस रोग से पीड़ित होते हैं।.. लेकिन यदि संयोगवश इन बच्चों को जीवन में यह रोग नहीं होता है तो सूत्र टूट जाता है एवं मूल प्रकंपक के पोते-पोती/नाती-नतिनी एवं परपोते-परपोती/परनाती-परनतिनी इस बात के प्रति निश्चित रहते हैं कि वे रोग से मुक्त हैं".[65][66] सर विलियम ऑस्लर की इस विकार में एवं सामान्य रूप से कोरिया में अभिरूचि थी, एवं वे हनटिंग्टन के दस्तावेज़ से यह कहते हुए प्रभावित हुए कि “चिकित्साशास्त्र के इतिहास में, इस बात के बहुत ही कम उदाहरण हैं जहां एक रोग का अधिक सटीक, अधिक सजीव या अधिक संक्षिप्त रूप से वर्णन किया गया है .“[62][67] HD में ऑस्लर की निरंतर अभिरूचि ने चिकित्साशास्त्र के क्षेत्र में उनके प्रभाव के साथ मिल कर, सम्पूर्ण चिकित्सा समुदाय में इस विकार के प्रति जागरूकता एवं ज्ञान के तेजी से प्रचार में मदद की.[62] लुइस थेयोफिल जोसेफ़ लैंडाउज़ी, डिज़ायर-मैग्लॉयर बोर्नविले, कैमिलो गोल्गी एवं जोसेफ़ जूल्स डेजेरिन सहित यूरोप में वैज्ञानिकों द्वारा अत्यधिक अभिरूचि दिखाई गई, एवं सदी के अंत तक, HD के संबंध में संपन्न अधिकांश शोध यूरोपीय मूल की थी।[62] 19वीं सदी के अंत तक, HD के संबंध में शोध एवं रिपोर्ट कई देशों में प्रकाशित हो चुके थे एवं रोग की पहचान एक विश्वव्यापी स्थिति के रूप में की जा चुकी थी।[62]
20वीं सदी में मेंडेल के वंशानुक्रम के पुनरन्वेषण के दौरान, HD का प्रयोग अस्थायी तौर पर एक अलिंगसूत्री प्रबल वंशानुक्रम के रूप में किया गया.[62] अंग्रेजी जीव-विज्ञानी विलियम बेटसन ने प्रभावित परिवार की वंशावली का यह पता लगाने के लिए प्रयोग किया कि HD का एक अलिंगसूत्र प्रबल वंशानुक्रम स्वरूप था।[63] मज़बूत वंशानुक्रम स्वरूप ने कई शोधकर्ताओं को पिछले अध्ययनों में शामिल परिवार के सदस्यों का पता लगाने एवं उन्हें जोड़ने का प्रयास करने के लिए प्रेरित किया, जिनमें से एक थे स्मिथ एली जेल्लिफ़.[62] जेल्लिफ़ ने संपूर्ण न्यूयॉर्क राज्य से जानकारी एकत्रित की एवं न्यू इंग्लैंड में HD की वंशावली के संबंध में कई लेख प्रकाशित किए.[68] जेल्लिफ़ के शोध ने उनके कॉलेज के मित्र चार्ल्स डेवनपोर्ट में अभिरूचि जगाई, जिन्होंने एलिज़ाबेथ मंकी को संयुक्त राज्य अमेरिका के पूर्वी तट पर HD ग्रस्त परिवारों के प्रथम क्षेत्रीय अध्ययन प्रस्तुत करने एवं उनकी वंशावली का निर्माण करने के लिए नियुक्त किया।[69] डेवनपोर्ट ने इस जानकारी का उपयोग HD के रोगलक्षणों की शुरूआत के विभिन्न उम्र एवं सीमा का प्रमाण प्रस्तुत करने एवं यह दावा करने के लिए किया कि संयुक्त राज्य अमेरिका में HD के अधिकांश मामले मुट्ठी भर व्यक्तियों में पाए जा सकते हैं।[69] इस शोध को 1932 में पी.आर.वेस्सी द्वारा और अधिक संवारा गया जिन्होंने इस विचार को लोकप्रिय बनाया कि 1630 में इंग्लैंड को छोड़ कर बॉस्टन जानेवाले तीन भाई संयुक्त राज्य अमेरिका में HD के जनक थे।[70] सबसे प्रारंभिक जनक मंकी, डेवनपोर्ट एवं वेस्सी के कार्यों से सुस्थापित एवं सुजनन संबंधी पूर्वाग्रह के दावे ने HD के संबंध में भ्रम एवं पूर्वाग्रह में योगदान दिया.[63] मंकी एवं डेवनपोर्ट ने इस विचार को भी लोकप्रिय बनाया कि प्राचीन काल में HD से प्रभावित कुछ व्यक्तियों को आत्माओं या जादू टोना के शिकार व्यक्तियों के नियंत्रणाधीन माना गया होगा एवं कभी-कभी वे समाज द्वारा त्यक्त या निष्कासित व्यक्ति होते थे।[71][72] इस विचार को सिद्ध नहीं किया गया और उदाहरण के लिए, इसके विपरीत कुछ प्रमाण मौजूद हैं कि जॉर्ज हनटिंग्टन द्वारा अध्ययन किए गए पारिवारिक समुदाय ने उन लोगों को मुक्त रूप से समायोजित किया जिन्होंने HD के रोगलक्षणों को प्रदर्शित किया।[63][71]
विकार के संबंध में शोध व्यवस्थित रूप से 20वीं सदी में जारी रहा, जिसने 1983 में एक प्रमुख महत्वपूर्ण खोज हासिल की जब संयुक्त राज्य अमेरिका-वेनेज़ुएला हनटिंग्टन रोग सहयोगी अनुसंधान परियोजना ने एक आकस्मिक जीन के सन्निकट स्थान का पता लगाया.[59] यह 1979 में शुरू किए गए एक विस्तृत अध्ययन का परिणाम था, जिसने दो पृथक वेनेज़ुएलीय गांवों, यथा बैरैंक्विटास एवं लैगुनेटास पर ध्यान केंद्रित किया जहां असामान्य रूप से इस रोग का अत्यधिक प्रचलन था। अन्य नवोन्मेषों में, परियोजना ने DNA चिह्नांकन विधियों का विकास किया जो मानव जीनोम परियोजना को संभव बनाने में एक महत्वपूर्ण क़दम था।[73] 1993 में शोध समूह ने वास्तविक आकस्मिक जीन को 4p16.3[74]पर पृथक किया, जिसने इसे आनुवंशिक सहलग्नता विश्लेषण का उपयोग करते हुए हासिल प्रथम अलिंगसूत्र रोग अवस्थिति बनाया.[74][75] उसी समय-सीमा में, जीन की लंबाई के प्रभावों से संबंधित अनीता हार्डिंग के शोध समूह के निष्कर्ष सहित, विकार की क्रियाविधि के संबंध में प्रमुख खोज किए जा रहे थे।[76]
विभिन्न प्रकार के पशुओं में इस रोग के नमूने तैयार करना, जैसे कि 1996 में विकसित परा-उत्पत्तिमूलक चूहे ने बड़े पैमाने पर किए जाने वाले प्रयोगों को संभव बनाया. इन पशुओं में मनुष्यों की तुलना में अधिक तेजी से होने वाले चयापचय और अधिक छोटे जीवन-काल के कारण, प्रयोगों से परिणाम अधिक शीघ्रता से प्राप्त होते हैं जो शोध में तेजी लाते हैं।[77][78] 1997 में mHtt खंडों के ग़लत बिखराव ने उनके द्वारा उत्पन्न किए जाने वाले नाभिकीय सम्मिलनों का पता लगाया.[79] इन प्रगतियों ने रोग में शामिल प्रोटीन, संभाव्य औषधि उपचार, देखभाल की विधियों एवं स्वयं जीन के संबंध में उत्तरोत्तर विस्तृत शोध को जन्म दिया.[62][80][81]
समाज और संस्कृति
नैतिकता
हनटिंग्टन रोग, विशेष रूप से बीमारी के लिए आनुवंशिक परीक्षण के प्रयोग ने कई नैतिक मुद्दों को उठाया है। आनुवंशिक परीक्षण के मुद्दों में ये परिभाषाएं शामिल है कि एक व्यक्ति को परीक्षण के लिए पात्र होने से पहले कितना परिपक्व होना चाहिए, परीक्षण की गोपनीयता कैसे सुनिश्चित होगी और रोजगार, जीवन बीमा या अन्य वित्तीय मामलों पर निर्णय के लिए कंपनियों को परीक्षण के परिणाम के उपयोग की अनुमति दी जानी चाहिए या नहीं. विवाद तब उठा था जब 1910 में चार्ल्स डेवनपोर्ट ने प्रस्तावित किया कि HD सहित कुछ रोगों से पीड़ित लोगों का अनिवार्य बंध्याकरण और आव्रजन नियंत्रण सुजननिक आंदोलन के हिस्से के रूप में किया जाना चाहिए.[82] कृत्रिम निषेचन में भ्रूणों के प्रयोग से संबंधित कुछ मुद्दे हैं। कुछ HD अनुसंधानों में अपने पशु परीक्षण और भ्रूणीय वंश कोशिकाओं के प्रयोग के कारण नैतिक मुद्दे भी है।[83][84]
हनटिंग्टन रोग के लिए एक सटीक निदान परीक्षण के विकास ने व्यक्ति के परिणामों के प्रयोग और पहुंच को लेकर सामाजिक, कानूनी और नैतिक चिंताएं उत्पन्न की हैं।[85][86] कई दिशा-निर्देश और परीक्षण प्रक्रियाएं प्रकटीकरण और गोपनीयता के लिए सख्त पद्धतियों का प्रयोग करती हैं ताकि यह तय किया जा सके कि कब और कैसे व्यक्तियों को अपने परिणाम प्राप्त करने के लिए अनुमत किया जा सके और ये परिणाम किन्हें उपलब्ध कराने हैं।[12] वित्तीय संस्थान और व्यवसाय इस सवाल का सामना कर रहे हैं किसी व्यक्ति के जीवन बीमा या रोजगार के लिए आकलन हेतु आनुवंशिक परीक्षण के परिणाम का उपयोग करे या नहीं. ब्रिटेन की बीमा कंपनियों ने इस बात पर सहमति जताई है कि 2014 तक वे इस जानकारी का उपयोग बीमा लेखन के अधिकांश मामलों में नहीं करेंगे.[87] बाद में शुरुआत की संभावनाओं के साथ अन्य लाइलाज आनुवंशिक स्थितियों के रूप में, यह नैतिकता की दृष्टि से आपत्तिजनक होगा कि एक बच्चे या किशोर पर पूर्व रोगसूचक परीक्षण किये जाएं क्योंकि उसे चिकित्सा लाभ नहीं मिलेगा.[26][88][89] केवल उन व्यक्तियों के परीक्षण के लिए आम सहमति है, जिन्हें ज्ञान के लिहाज से परिपक्व माना जाता है, यद्यपि एक जवाबी तर्क है कि माता-पिता को अपने बच्चे की ओर से निर्णय करने का पूरा अधिकार है।[26][88][89] प्रभावी उपचार के अभाव में, कानूनी उम्र से कम व्यक्ति का परीक्षण सक्षम नहीं माना जाता है, इसे ज्यादातर मामलों में अनैतिक माना जाता है।[26][88][89]
यह सुनिश्चित करने के लिए कि बच्चे को कोई विशिष्ट रोग नहीं है, प्रसवपूर्व आनुवंशिक परीक्षण या रोपणपूर्व आनुवंशिक रोगनिदान कुछ नैतिक चिंताएं पैदा करती हैं।[90] उदाहरण के लिए, प्रसवपूर्व परीक्षण चयनात्मक गर्भपात का मुद्दा उठाता है, जो कई लोगो को अस्वीकार्य है।[90] HD के लिए रोपणपूर्व परीक्षण के उपयोग हेतु कृत्रिम निषेचन के लिए प्रयुक्त भ्रूणों की संख्या से दुगुने भ्रूणों की जरूरत होती है, क्योंकि इनमे से आधे HD के लिए सकारात्मक होंगे. एक प्रबल बीमारी के लिए ऐसी परिस्थियों में कठिनाइयां उत्पन्न हो सकती है जहां माता-पिता अपने स्वयं के निदान को जानना नहीं चाहते हैं, क्योंकि इसमें माता-पिता से प्रक्रिया के कुछ हिस्सों को गुप्त रखा जाएगा.[90]
सहायता संगठन


1968 में, अपनी पत्नी के परिवार में HD का पता लगने के बाद, डॉ॰ मिल्टन वेक्स्लर, अनुसंधान के समन्वय और समर्थन से आनुवंशिक बीमारियों का इलाज करने के उद्देश्य से वंशानुगत रोग संस्थान (HDF) प्रारंभ करने के लिए प्रेरित हुए थे।[91] संस्थान और डॉ॰वेक्स्लर की बेटी नैन्सी वेक्स्लर, वेनेज़ुएला के उस अनुसंधान दल के मुख्य अंश थे जिसने HD जीन की खोज की थी।[91] जब HDF का गठन हुआ, तो लगभग उसी समय मार्जोरी गुथरी ने अपने पति वूडी गुथरी की HD की समस्याओं से मौत के बाद हनटिंग्टन रोग से जूझने के लिए एक समिति (अब हनटिंग्टन्स डिज़ीस सोसाइटी ऑफ़ अमेरिका) के गठन में मदद की.[92] तब से लेकर अब तक, विश्व भर के कई देशों में समर्थन और अनुसंधान संगठनों का गठन हुआ है, जिन्होंने HD के प्रति सार्वजनिक जागरूकता बढ़ाने में मदद की है। इनमें से अनेक, अंतर्राष्ट्रीय हनटिंग्टन संस्था और यूरो HD नेटवर्क जैसे संगठनों के साथ सहयोग करते हैं।[93] कई सहायता संगठन वार्षिक HD जागरूकता कार्यक्रम रखते हैं, जिनमें से कुछ उनकी संबद्ध सरकारों द्वारा समर्थित हैं। उदाहरण के लिए, अमेरिका की सीनेट ने 6 जून को राष्ट्रीय "हनटिंग्टन रोग जागरूकता दिवस" घोषित किया है।[94]
अनुसंधान संबंधी निर्देश
HD की प्रक्रिया के अनुसंधान में Htt की कार्यप्रणाली को पहचानने, उससे किस प्रकार mHtt भिन्न है या उसमें दखल देती है, तथा उसके द्वारा उत्पन्न मस्तिष्क विकृति पर ध्यान केंद्रित किया जाता है।[25] अधिकांश अनुसंधान पशुओं पर किए जाते हैं। उपयुक्त पशु मॉडल, रोग पैदा करने की मूल प्रक्रिया को समझने और दवा के विकास के प्रारंभिक दौर की सहायता के लिए महत्वपूर्ण हैं।[95] रासायनिक प्रेरण द्वारा HD-सदृश लक्षणों को दर्शाने के लिए चूहों और बंदरों का प्रयोग किया गया था,[95][96][97] लेकिन उन्होंने रोग के प्रारंभिक लक्षणों की नक़ल नहीं दर्शाई. हनटिंग्टन जीन की पहचान के बाद से, HD-सदृश संलक्षणों को दर्शाने वाले ट्रांस्जेनिक प्राणियों (चूहों,[95][98][99] ड्रोसोफिला फलों की मक्खियों,[95][100] और हाल ही में बंदरों[101]) को जीन में CAG आवृत्ति व्याप्ति द्वारा उत्पन्न किया जा सकता है। जब जीन प्रकट हो रहा हो तो निमेटोड कीड़े भी एक मूल्यवान मॉडल प्रदान करते हैं।[95][102]
इंट्राबाडीज़ कहलाने वाले आनुवंशिक रूप से निर्मित अंतर्कोशिकीय प्रतिरक्षी खंड ड्रोसोफिला मॉडल के शुरूआती चरण के दौरान मृत्यु दर को रोकने में सफल दिखते हैं। उनकी क्रिया विधि mHtt संग्रहण के लिए निषेध थी।[95][103][104] क्योंकि HD निर्णायक रूप से एकल जीन से जुड़ा है, जीन विस्मृति सशक्त रूप से संभावित है और चूहों के मॉडल में जीन पछाड़ के प्रयोग से, शोधकर्ताओं ने दिखाया है कि जब mHtt का प्रभाव कम हो जाता है तो लक्षणों में सुधार होता है।[46][105][106] वंश कोशिका उपचार में, मस्तिष्क के प्रभावित हिस्सों में वंश कोशिका के प्रत्यारोपण से क्षतिग्रस्त न्यूरॉनों को बदल दिया जाता है। पशुओं के मॉडल पर और प्रारंभिक मानव नैदानिक परीक्षणों में इस तकनीक के प्रयोगों से कुछ सकारात्मक परिणाम मिले हैं।[107]
पशुओं में अनेक दवाएं लाभदायक परिणाम उत्पन्न करने की रिपोर्टें आई हैं, जिनमें शामिल हैं क्रिएटीन, सह-एन्ज़ाइम Q10 और एंटीबायोटिक मिनोसाइक्लीन.[46] बाद में इनमें से कुछ की मानवों द्वारा चिकित्सकीय परीक्षण में जांच की गई हैं और यथा 2009 इनमें से कई परीक्षणों के विभिन्न चरणों में हैं।[46]
सन्दर्भ
- ↑ अ आ इ ई उ ऊ ए ऐ Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 218. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ अ आ इ "Huntington Disease". genereviews bookshelf. University of Washington. 2007-07-19. मूल से 2 जून 2010 को पुरालेखित. अभिगमन तिथि 2009-03-12.
- ↑ अ आ Kremer B (2002). "Clinical neurology of Huntington's disease". प्रकाशित Bates G, Harper P, and Jones L (संपा॰). Huntington's Disease – Third Edition. Oxford: Oxford University Press. पपृ॰ 28–53. आई॰ऍस॰बी॰ऍन॰ 0-19-851060-8.सीएस1 रखरखाव: एक से अधिक नाम: editors list (link)
- ↑ Wagle, A C (2000). "Psychiatric Morbidity in Huntington's disease". Neurology, Psychiatry and Brain Research (8): 5–16. नामालूम प्राचल
|coauthors=की उपेक्षा की गयी (|author=सुझावित है) (मदद) - ↑ अ आ इ ई उ ऊ ए ऐ Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 219. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ अ आ इ ई उ ऊ ए ऐ Montoya A, Price BH, Menear M, Lepage M (2006). "Brain imaging and cognitive dysfunctions in Huntington's disease" (PDF). J Psychiatry Neurosci. 31 (1): 21–9. PMID 16496032. पी॰एम॰सी॰ 1325063. मूल (PDF) से 23 मार्च 2016 को पुरालेखित. अभिगमन तिथि 2009-04-01.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Aziz NA, van der Marck MA, Pijl H, Olde Rikkert MG, Bloem BR, Roos RA (2008). "Weight loss in neurodegenerative disorders". J. Neurol. 255 (12): 1872–80. PMID 19165531. डीओआइ:10.1007/s00415-009-0062-8.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ "Booklet by the Huntington Society of Canada" (PDF). Caregiver's Handbook for Advanced-Stage Huntington Disease. HD Society of Canada. 2007-04-11. मूल (PDF) से 25 जून 2008 को पुरालेखित. अभिगमन तिथि 2008-08-10.
- ↑ Gagnon JF, Petit D, Latreille V, Montplaisir J (2008). "Neurobiology of sleep disturbances in neurodegenerative disorders". Curr. Pharm. Des. 14 (32): 3430–45. PMID 19075719. डीओआइ:10.2174/138161208786549353.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ ई van Duijn E, Kingma EM, van der Mast RC (2007). "Psychopathology in verified Huntington's disease gene carriers". J Neuropsychiatry Clin Neurosci. 19 (4): 441–8. PMID 18070848. डीओआइ:10.1176/appi.neuropsych.19.4.441.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ वैन डर बर्ग जेएम, जोर्क्विस्ट एम, ब्रंडीन पी. (2009). बियॉन्ड द ब्रेन: वाइडस्प्रेड पेथॉलोजी इन हनटिंग्टन्स डिज़ीस. लैनसेट न्यूरोल. 8(8):765-74. doi:10.1016/S1474-4422(09)70178-4पमिड 19608102
- ↑ अ आ इ ई उ ऊ ए ऐ ओ औ क ख ग घ ङ च छ ज झ ञ Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 221. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ अ आ इ ई उ ऊ ए ऐ ओ औ क ख ग घ ङ च छ ज झ ञ ट ठ ड Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 220. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ Katsuno M, Banno H, Suzuki K; एवं अन्य (2008). "Molecular genetics and biomarkers of polyglutamine diseases". Curr. Mol. Med. 8 (3): 221–34. PMID 18473821. डीओआइ:10.2174/156652408784221298. मूल से 13 फ़रवरी 2009 को पुरालेखित. अभिगमन तिथि 2009-04-01. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ अ आ Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 222. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ Nance MA, Myers RH (2001). "Juvenile onset Huntington's disease—clinical and research perspectives". Ment Retard Dev Disabil Res Rev. 7 (3): 153–7. PMID 11553930. डीओआइ:10.1002/mrdd.1022.
- ↑ अ आ Passarge, E (2001). Color Atlas of Genetics (2nd संस्करण). Thieme. पृ॰ 142. आई॰ऍस॰बी॰ऍन॰ 0-86577-958-9.
- ↑ Ridley RM, Frith CD, Crow TJ, Conneally PM (1988). "Anticipation in Huntington's disease is inherited through the male line but may originate in the female". Journal of Medical Genetics. 25 (9): 589–595. PMID 2972838. डीओआइ:10.1136/jmg.25.9.589. पी॰एम॰सी॰ 1051535. मूल से 14 जुलाई 2012 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Semaka A, Creighton S, Warby S, Hayden MR (2006). "Predictive testing for Huntington disease: interpretation and significance of intermediate alleles". Clin. Genet. 70 (4): 283–94. PMID 16965319. डीओआइ:10.1111/j.1399-0004.2006.00668.x.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ Squitieri F, Gellera C, Cannella M; एवं अन्य (2003). "Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course". Brain. 126 (Pt 4): 946–55. PMID 12615650. डीओआइ:10.1093/brain/awg077. मूल से 20 दिसंबर 2012 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Wexler NS, Young AB, Tanzi RE; एवं अन्य (1987). "Homozygotes for Huntington's disease". Nature. 326 (6109): 194–197. PMID 2881213. डीओआइ:10.1038/326194a0. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Goehler H, Lalowski M, Stelzl U; एवं अन्य (2004). "A protein interaction network links GIT1, an enhancer of Huntingtin aggregation, to Huntington's disease". Mol. Cell. 15 (6): 853–65. PMID 15383276. डीओआइ:10.1016/j.molcel.2004.09.016. मूल से 5 अक्तूबर 2017 को पुरालेखित. अभिगमन तिथि 2009-04-27. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Harjes P, Wanker EE (2003). "The hunt for huntingtin function: interaction partners tell many different stories". Trends Biochem. Sci. 28 (8): 425–33. PMID 12932731. डीओआइ:10.1016/S0968-0004(03)00168-3. मूल से 5 अक्तूबर 2017 को पुरालेखित. अभिगमन तिथि 2009-04-27.
- ↑ अ आ इ Cattaneo E, Zuccato C, Tartari M (2005). "Normal huntingtin function: an alternative approach to Huntington's disease". Nat. Rev. Neurosci. 6 (12): 919–30. PMID 16288298. डीओआइ:10.1038/nrn1806.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ ई उ ऊ Rubinsztein DC, Carmichael J (2003). "Huntington's disease: Molecular basis of neurodegeneration". Expert Rev Mol Med. 5 (20): 1–21. PMID 14585171. डीओआइ:10.1017/S1462399403006549.
- ↑ अ आ इ ई Bloch M, Hayden MR (1990). "Opinion: predictive testing for Huntington disease in childhood: challenges and implications". Am. J. Hum. Genet. 46 (1): 1–4. PMID 2136787. पी॰एम॰सी॰ 1683548.
- ↑ अ आ इ "Huntingtin Protein and Protein Aggregation | HOPES - A guide to the science of Huntington's disease". मूल से 31 अगस्त 2010 को पुरालेखित.
- ↑ अ आ Subramaniam S, Sixt KM, Barrow R, Snyder SH (2009). "Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity". Science. 324 (5932): 1327–30. PMID 19498170. डीओआइ:10.1126/science.1172871. पी॰एम॰सी॰ 2745286.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ "The Basic Neurobiology of Huntington's Disease (Text and Audio) | HOPES - A guide to the science of Huntington's disease". मूल से 20 नवंबर 2010 को पुरालेखित.
- ↑ अ आ इ ई "Nature Clinical Practice Neurology | Mechanisms of Disease: histone modifications in Huntington's disease | Article". मूल से 26 नवंबर 2011 को पुरालेखित.
- ↑ Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia A-S, McNamara JO, Williams SM (2001). "Modulation of Movement by the Basal Ganglia – Circuits within the Basal Ganglia System". प्रकाशित Purves D (संपा॰). Neuroscience (2nd संस्करण). Sunderland, MA: Sinauer Associates. आई॰ऍस॰बी॰ऍन॰ 0-87893-742-0. मूल से 17 अगस्त 2009 को पुरालेखित. अभिगमन तिथि 2009-04-01.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Lobsiger CS, Cleveland DW (2007). "Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease". Nat. Neurosci. 10 (11): 1355–60. PMID 17965655. डीओआइ:10.1038/nn1988.
- ↑ अ आ Crossman AR (2000). "Functional anatomy of movement disorders" (PDF). J. Anat. 196 (Pt 4): 519–25. PMID 10923984. डीओआइ:10.1046/j.1469-7580.2000.19640519.x. पी॰एम॰सी॰ 1468094.[मृत कड़ियाँ]
- ↑ अ आ "Analysis of Strand Slippage in DNA Polymerase Expansions of CAG/CTG Triplet Repeats Associated with Neurodegenerative Disease — JBC". मूल से 8 सितंबर 2017 को पुरालेखित.
- ↑ "Unified Huntington's Disease Rating Scale (UHDRS)". UHDRS and Database. HSG. 2009-02-01. मूल से 2 मई 2009 को पुरालेखित. अभिगमन तिथि 2009-04-14.
- ↑ Myers RH (2004). "Huntington's disease genetics". NeuroRx. 1 (2): 255–62. PMID 15717026. डीओआइ:10.1602/neurorx.1.2.255. पी॰एम॰सी॰ 534940.
- ↑ Burson CM, Markey KR (2001). "Genetic counseling issues in predictive genetic testing for familial adult-onset neurologic diseases". Semin Pediatr Neurol. 8 (3): 177–86. PMID 11575847. डीओआइ:10.1053/spen.2001.26451.
- ↑ अ आ Hayden MR (2003). "Predictive testing for Huntington's disease: a universal model?". Lancet Neurol. 2 (3): 141–2. PMID 12849232. डीओआइ:10.1016/S1474-4422(03)00317-X. नामालूम प्राचल
|month=की उपेक्षा की गयी (मदद) - ↑ "Guidelines for the molecular genetics predictive test in Huntington's disease. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington's Chorea". Neurology. 44 (8): 1533–6. 1994. PMID 8058167.
- ↑ Kuliev A, Verlinsky Y (2005). "Preimplantation diagnosis: A realistic option for assisted reproduction and genetic practice". Curr. Opin. Obstet. Gynecol. 17 (2): 179–83. PMID 15758612. डीओआइ:10.1097/01.gco.0000162189.76349.c5. मूल से 13 अगस्त 2011 को पुरालेखित. अभिगमन तिथि 2009-04-01.
- ↑ अ आ इ ई उ Schneider SA, Walker RH, Bhatia KP (2007). "The Huntington's disease-like syndromes: what to consider in patients with a negative Huntington's disease gene test". Nat Clin Pract Neurol. 3 (9): 517–25. PMID 17805246. डीओआइ:10.1038/ncpneuro0606.
|access-date=दिए जाने पर|url= भी दिया जाना चाहिए(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Frank S, Jankovic J. (2010). "Advances in the Pharmacological Management of Huntington's Disease". Drugs. 70 (5): 561–71. PMID 20329804. डीओआइ:10.2165/11534430-000000000-00000. मूल से 8 अक्तूबर 2011 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.
- ↑ अ आ इ ई उ ऊ ए Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 224. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ अ आ इ Bonelli RM, Wenning GK, Kapfhammer HP (2004). "Huntington's disease: present treatments and future therapeutic modalities". Int Clin Psychopharmacol. 19 (2): 51–62. PMID 15076012. डीओआइ:10.1097/00004850-200403000-00001. मूल से 1 जून 2012 को पुरालेखित. अभिगमन तिथि 2009-04-01.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ "FDA Approves First Drug for Treatment of Chorea in Huntington's Disease". FDA Approves First Drug for Treatment of Chorea in Huntington's Disease. U.S. Food and Drug Administration. अगस्त 15, 2008. मूल से 1 जून 2012 को पुरालेखित. अभिगमन तिथि 2008-08-10.
- ↑ अ आ इ ई Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 225. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ Panagiotakis PH, DiSario JA, Hilden K, Ogara M, Fang JC (2008). "DPEJ tube placement prevents aspiration pneumonia in high-risk patients". Nutr Clin Pract. 23 (2): 172–5. PMID 18390785. डीओआइ:10.1177/0884533608314537.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)[मृत कड़ियाँ]
- ↑ Bilney B, Morris ME, Perry A (2003). "Effectiveness of physiotherapy, occupational therapy, and speech pathology for people with Huntington's disease: a systematic review". Neurorehabil Neural Repair. 17 (1): 12–24. PMID 12645441. डीओआइ:10.1177/0888439002250448.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)[मृत कड़ियाँ]
- ↑ Zinzi P, Salmaso D, De Grandis R; एवं अन्य (2007). "Effects of an intensive rehabilitation programme on patients with Huntington's disease: a pilot study". Clin Rehabil. 21 (7): 603–13. PMID 17702702. डीओआइ:10.1177/0269215507075495. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)[मृत कड़ियाँ] - ↑ Harper P (2002). "Genetic counselling and presymptomatic testing". प्रकाशित Bates G, Harper P, and Jones L (संपा॰). Huntington's Disease – Third Edition. Oxford: Oxford University Press. पपृ॰ 198–242. आई॰ऍस॰बी॰ऍन॰ 0-19-851060-8.सीएस1 रखरखाव: एक से अधिक नाम: editors list (link)
- ↑ Harper PS (1999). "Huntington's disease: A clinical, genetic and molecular model for polyglutamine repeat disorders". Philos. Trans. R. Soc. Lond., B, Biol. Sci. 354 (1386): 957–61. PMID 10434293. डीओआइ:10.1098/rstb.1999.0446. पी॰एम॰सी॰ 1692597.
- ↑ Andrew SE, Goldberg YP, Kremer B; एवं अन्य (1993). "The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease". Nat. Genet. 4 (4): 398–403. PMID 8401589. डीओआइ:10.1038/ng0893-398. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Crauford D and Snowden J (2002). "Neuropyschological and neuropsychiatric aspects of Huntington's disease". प्रकाशित Bates G, Harper P, and Jones L (संपा॰). Huntington's Disease – Third Edition. Oxford: Oxford University Press. पपृ॰ 62–87. आई॰ऍस॰बी॰ऍन॰ 0-19-851060-8.सीएस1 रखरखाव: एक से अधिक नाम: editors list (link)
- ↑ Di Maio L, Squitieri F, Napolitano G; एवं अन्य (1993). "Suicide risk in Huntington's disease". J. Med. Genet. 30 (4): 293–5. PMID 8487273. डीओआइ:10.1136/jmg.30.4.293. पी॰एम॰सी॰ 1016335. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ अ आ इ ई उ ऊ Harper P (2002). "The epidemiology of Huntington's disease". प्रकाशित Bates G, Harper P, and Jones L (संपा॰). Huntington's Disease – Third Edition. Oxford: Oxford University Press. पपृ॰ 159–189. आई॰ऍस॰बी॰ऍन॰ 0-19-851060-8.सीएस1 रखरखाव: एक से अधिक नाम: editors list (link)
- ↑ Sharon I (2010). "Huntington Disease Dementia". emedicine, WebMD. Medscape. मूल से 5 मार्च 2010 को पुरालेखित. अभिगमन तिथि 2010-05-16. नामालूम प्राचल
|coauthors=की उपेक्षा की गयी (|author=सुझावित है) (मदद) - ↑ Driver-Dunckley E, Caviness JN. (2007). "Huntington's disease". प्रकाशित Schapira AHV (संपा॰). Neurology and Clinical Neuroscience. Mosby Elsevier. पपृ॰ 879–885. आई॰ऍस॰बी॰ऍन॰ 978-0323033541.
- ↑ Avila-Giron R. (1973). "Medical and Social Aspects of Huntington's chorea in the state of Zulia, Venezuela". Advances in Neurology. 1: 261–6.
- ↑ अ आ Gusella JF, Wexler NS, Conneally PM; एवं अन्य (1983). "A polymorphic DNA marker genetically linked to Huntington's disease". Nature. 306 (5940): 234–8. PMID 6316146. डीओआइ:10.1038/306234a0. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Squitieri F, Andrew SE, Goldberg YP; एवं अन्य (1994). "DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence". Hum. Mol. Genet. 3 (12): 2103–14. PMID 7881406. डीओआइ:10.1093/hmg/3.12.2103. मूल से 15 मार्च 2020 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Almqvist EW, Elterman DS, MacLeod PM, Hayden MR (2001). "High incidence rate and absent family histories in one quarter of patients newly diagnosed with Huntington disease in British Columbia". Clin. Genet. 60 (3): 198–205. PMID 11595021. डीओआइ:10.1034/j.1399-0004.2001.600305.x.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ ई उ ऊ ए ऐ ओ सन्दर्भ त्रुटि:
<ref>का गलत प्रयोग;OxfordMonographHistoryनाम के संदर्भ में जानकारी नहीं है। - ↑ अ आ इ ई Wexler A, Wexler N (2008). The Woman Who Walked Into the Sea. Huntington's and the Making of a Genetic Disease. Yale University Press. पृ॰ 288. आई॰ऍस॰बी॰ऍन॰ 9780300105025. मूल से 6 जून 2011 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.
- ↑ Lund JC (1860). "Chorea Sti Viti i Sætersdalen. Uddrag af Distriktslæge J. C. Lunds Medicinalberetning for 1860". Beretning om Sundhedstilstanden. Norway: 137–138.
- ↑ सन्दर्भ त्रुटि:
<ref>का गलत प्रयोग;onchoreaनाम के संदर्भ में जानकारी नहीं है। - ↑ Lanska DJ (2000). "George Huntington (1850–1916) and hereditary chorea". J Hist Neurosci. 9 (1): 76–89. PMID 11232352. डीओआइ:10.1076/0964-704X(200004)9:1;1-2;FT076.
- ↑ Irwin A Brody, Robert H Wilkins (1967). "Huntington's Chorea". Arch Neurol. 17 (3): 331. PMID 4228262. डीओआइ:10.1001/archneur.1967.00470270109013. मूल से 8 मार्च 2012 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.
- ↑ Jelliffe SE, Muncey EB, Davenport CB (1913). "Huntington's Chorea: A Study in Heredity". The Journal of Nervous and Mental Disease. 40 (12): 796. मूल से 12 मई 2012 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ Davenport CB, Muncey EB (1916). "Huntington's chorea in relation to heredity and eugenics". American Journal of Insanity. 73: 195–222. डीओआइ:10.1176/appi.ajp.73.2.195. मूल से 11 जून 2011 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.
- ↑ Vessie, PR (1932). "On the transmission of Huntington's chorea for 300 years – the Bures family group". Nervous and Mental Disease. Baltimore. 76 (6): 553–573. डीओआइ:10.1097/00005053-193212000-00001. अभिगमन तिथि 2009-04-01.
- ↑ अ आ Wexler AR (2002). "Chorea and community in a 19th-century town". Bull Hist Med. 76 (3): 495–527. PMID 12486915. डीओआइ:10.1353/bhm.2002.0150.
- ↑ Conneally PM (1984). "Huntington disease: genetics and epidemiology". Am. J. Hum. Genet. 36 (3): 506–26. PMID 6233902. पी॰एम॰सी॰ 1684448.
- ↑ "The Venezuela Huntington's disease project". Hereditary Disease Foundation website. Hereditary Disease Foundation. 2008. मूल से 26 अक्तूबर 2008 को पुरालेखित. अभिगमन तिथि 2008-09-08.
- ↑ अ आ Macdonald M (1993). "A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group". Cell. 72 (6): 971–83. PMID 8458085. डीओआइ:10.1016/0092-8674(93)90585-E. मूल से 5 अक्तूबर 2017 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.
- ↑ Bertram L, Tanzi RE (2005). "The genetic epidemiology of neurodegenerative disease". J. Clin. Invest. 115 (6): 1449–57. PMID 15931380. डीओआइ:10.1172/JCI24761. पी॰एम॰सी॰ 1137006.
|access-date=दिए जाने पर|url= भी दिया जाना चाहिए(मदद) - ↑ La Spada AR, Roling DB, Harding AE; एवं अन्य (1992). "Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy". Nat. Genet. 2 (4): 301–4. PMID 1303283. डीओआइ:10.1038/ng1292-301. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Sathasivam K, Hobbs C, Mangiarini L; एवं अन्य (1999). "Transgenic models of Huntington's disease". Philos. Trans. R. Soc. Lond., B, Biol. Sci. 354 (1386): 963–9. PMID 10434294. डीओआइ:10.1098/rstb.1999.0447. पी॰एम॰सी॰ 1692600. मूल से 23 फ़रवरी 2020 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Li JY, Popovic N, Brundin P (2005). "The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies". NeuroRx. 2 (3): 447–64. PMID 16389308. डीओआइ:10.1602/neurorx.2.3.447. पी॰एम॰सी॰ 1144488.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ DiFiglia M, Sapp E, Chase KO; एवं अन्य (1997). "Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain". Science. 277 (5334): 1990–3. PMID 9302293. डीओआइ:10.1126/science.277.5334.1990. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ "Achievements of Hereditary Disease Foundation". Achievements of Hereditary Disease Foundation. Hereditary Disease Foundation. मूल से 16 जुलाई 2011 को पुरालेखित. अभिगमन तिथि 2008-08-10.
- ↑ "HDA research news—medical research into treatment & prevention on hda.org.uk". Huntington's Disease Association-United Kingdom. 2009. मूल से 31 मई 2009 को पुरालेखित. अभिगमन तिथि 2008-08-10.
- ↑ Davenport CB (1915). "Huntington's chorea in relation to heredity and eugenics". Proc. Natl. Acad. Sci. U.S.A. 1 (5): 283–5. PMID 16575999. डीओआइ:10.1073/pnas.1.5.283. पी॰एम॰सी॰ 1090803.
- ↑ Rollin, Bernard E. (2006). "The Regulation of Animal Research and the Emergence of Animal Ethics: A Conceptual History". Theoretical Medicine and Bioethics. 27 (4): 285–304. PMID 16937023. डीओआइ:10.1007/s11017-006-9007-8.
- ↑ Doerflinger RM (2008). "The problem of deception in embryonic stem cell research". Cell Prolif. 41 Suppl 1: 65–70. PMID 18181947. डीओआइ:10.1111/j.1365-2184.2008.00492.x.
- ↑ Chapman MA (1990). "Predictive testing for adult-onset genetic disease: ethical and legal implications of the use of linkage analysis for Huntington disease". Am. J. Hum. Genet. 47 (1): 1–3. PMID 2140926. पी॰एम॰सी॰ 1683745.
- ↑ Huggins M, Bloch M, Kanani S; एवं अन्य (1990). "Ethical and legal dilemmas arising during predictive testing for adult-onset disease: the experience of Huntington disease". Am. J. Hum. Genet. 47 (1): 4–12. PMID 1971997. पी॰एम॰सी॰ 1683755. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ "BBC article: Genetic data banned for insurers". BBC. 2008-06-13. अभिगमन तिथि 2008-08-10.
- ↑ अ आ इ Binedell J, Soldan JR, Scourfield J, Harper PS (1996). "Huntington's disease predictive testing: the case for an assessment approach to requests from adolescents". J. Med. Genet. 33 (11): 912–8. PMID 8950670. डीओआइ:10.1136/jmg.33.11.912. पी॰एम॰सी॰ 1050784.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ Borry P, Goffin T, Nys H, Dierickx K (2008). "Predictive genetic testing in minors for adult-onset genetic diseases". Mt. Sinai J. Med. 75 (3): 287–96. PMID 18704981. डीओआइ:10.1002/msj.20038.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ इ Braude PR, De Wert GM, Evers-Kiebooms G, Pettigrew RA, Geraedts JP (1998). "Non-disclosure preimplantation genetic diagnosis for Huntington's disease: practical and ethical dilemmas". Prenat. Diagn. 18 (13): 1422–6. PMID 9949442. डीओआइ:10.1002/(SICI)1097-0223(199812)18:13<1422::AID-PD499>3.0.CO;2-R. अभिगमन तिथि 2009-04-01.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ अ आ "Hereditary Disease Foundation – About Us". Hereditary disease foundation. 2008. मूल से 12 मार्च 2009 को पुरालेखित. अभिगमन तिथि 2009-03-27.
- ↑ "Huntington's Disease Society of America – History". Huntington's Disease Society of America. 2008. मूल से 24 नवंबर 2010 को पुरालेखित. अभिगमन तिथि 2009-03-17.
- ↑ "IHA Profile". International Huntington Association. 2004. मूल से 12 जून 2011 को पुरालेखित. अभिगमन तिथि 2009-04-03.
- ↑ "US Senate s. resolution 531" (PDF). S. Res. 531. US Senate. 2008-04-06. मूल से 21 जुलाई 2011 को पुरालेखित (PDF). अभिगमन तिथि 2008-08-10.
- ↑ अ आ इ ई उ ऊ Walker FO (2007). "Huntington's disease". Lancet. 369 (9557): 223. PMID 17240289. डीओआइ:10.1016/S0140-6736(07)60111-1.
- ↑ Coyle JT, Schwarcz R (1976). "Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea". Nature. 263 (5574): 244–6. PMID 8731. डीओआइ:10.1038/263244a0.
- ↑ Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF (1995). "Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates". Proc Natl Acad Sci USA. 92 (15): 7105–7109. PMID 7624378. डीओआइ:10.1073/pnas.92.15.7105. पी॰एम॰सी॰ 41480.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996). "Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological genotype in transgenic mice". Cell. 87 (3): 493–506. PMID 8898202. डीओआइ:10.1016/S0092-8674(00)81369-0.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, and Morton AJ (15 अप्रैल 1999). "Characterization of Progressive Motor Deficits in Mice Transgenic for the Human Huntington's Disease Mutation". The Journal of Neuroscience. 19 (8): 3248–3257. PMID 10191337. मूल से 7 जनवरी 2009 को पुरालेखित. अभिगमन तिथि 2009-04-01.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Marsh JL, Pallos J and Thompson LM (2003). "Fly models of Huntington's disease". Human Molecular Genetics. 12 (2): 187–193. PMID 12925571. डीओआइ:10.1093/hmg/ddg271. मूल से 24 दिसंबर 2009 को पुरालेखित. अभिगमन तिथि 2009-04-01.
- ↑ "First Transgenic Monkey Model Of Huntington's Disease Developed". ScienceDaily. 2008-05-19. मूल से 5 जून 2011 को पुरालेखित. अभिगमन तिथि 2008-08-10.
- ↑ Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC (2007). "Identification of potential therapeutic drugs for huntington's disease using Caenorhabditis elegans". PLoS ONE. 2 (6): e504. PMID 17551584. डीओआइ:10.1371/journal.pone.0000504. पी॰एम॰सी॰ 1876812. मूल से 13 सितंबर 2011 को पुरालेखित. अभिगमन तिथि 15 जनवरी 2011.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- ↑ Lecerf JM, Shirley TL, Zhu Q; एवं अन्य (2001). "Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington's disease". Proc. Natl. Acad. Sci. U.S.A. 98 (8): 4764–9. PMID 11296304. डीओआइ:10.1073/pnas.071058398. पी॰एम॰सी॰ 31908. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Miller TW, Zhou C, Gines S; एवं अन्य (2005). "A human single-chain Fv intrabody preferentially targets amino-terminal huntingtin's fragments in striatal models of Huntington's disease". Neurobiol. Dis. 19 (1–2): 47–56. PMID 15837560. डीओआइ:10.1016/j.nbd.2004.11.003. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Harper SQ, Staber PD, He X; एवं अन्य (2005). "RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model". Proc. Natl. Acad. Sci. U.S.A. 102 (16): 5820–5. PMID 15811941. डीओआइ:10.1073/pnas.0501507102. पी॰एम॰सी॰ 556303. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Díaz-Hernández M, Torres-Peraza J, Salvatori-Abarca A; एवं अन्य (2005). "Full Motor Recovery Despite Striatal Neuron Loss and Formation of Irreversible Amyloid-Like Inclusions in a Conditional Mouse Model of Huntington's Disease". The Journal of Neuroscience. 25 (42): 9773–9781. PMID 16237181. डीओआइ:10.1523/JNEUROSCI.3183-05.2005. Explicit use of et al. in:
|author=(मदद)सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) - ↑ Clelland CD, Barker RA, Watts C (2008). "Cell therapy in Huntington disease". Neurosurg Focus. 24 (3–4): E9. PMID 18341412. डीओआइ:10.3171/FOC/2008/24/3-4/E8.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)